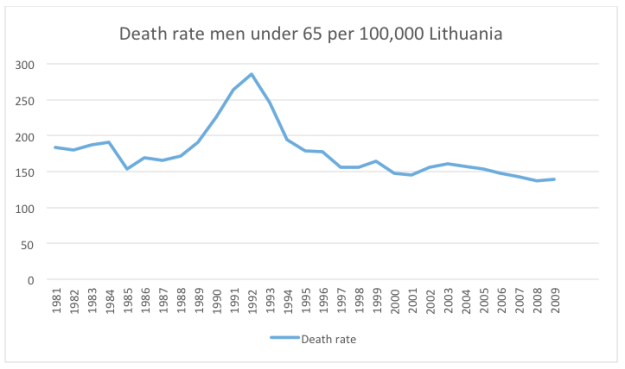
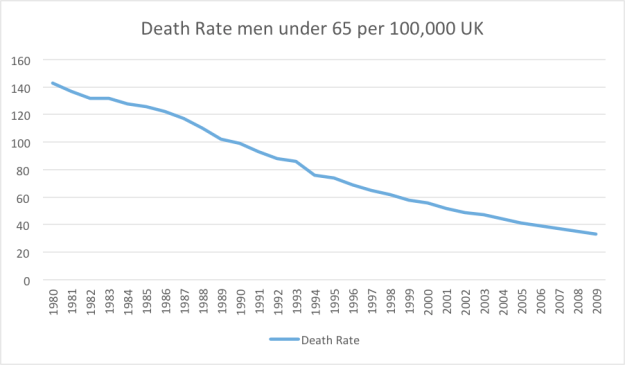
“If liberty means anything at all, it means the right to tell people what they do not want to hear.” George Orwell.
Many of you may be aware of an article published in the Lancet on the eighth of September. ‘Interpretation of the evidence for the efficacy and safety of statin therapy.’1 It caused a media stir, and I was asked to appear on a few BBC programmes to argue against it – tricky in two minutes. At one stage I was cut off when I attempted to bring up the issue of financial conflicts of interest amongst the authors. The lead author of this paper was Professor Sir Rory Collins.
In truth, I have been awaiting this article for some time. In fact, I am going to reproduce here a blog I wrote on February 16th 2015, predicting exactly what was going to happen, who was going to be involved, and (in broad terms) exactly what they were going to say:
A humiliating climb down – or a Machiavellian move?
Some of you may have seen a headline in the Sunday Express Newspaper ‘Statin, new safety checks.’ The subheading was ‘Oxford professor who championed controversial drug to reassess evidence of side effects.’
Those of you who read this blog probably know that the professor in question is Sir Rory Collins. He, more than anyone, has championed the ever wider prescription of these drugs. He has also ruthlessly attacked anyone who dares make any criticism of them.
You may remember that last year he tried to get the BMJ to retract two articles claiming that statins had side effects (correctly called adverse effects, but I will call them side-effects to avoid confusion) of around 18 – 20%.
He stated that these articles were irresponsible, worse than Andrew Wakefield’s work on the MMR vaccine, and that thousands would die if they were scared off taking their statins by such articles. Ah yes, the old ‘thousands will die’ game. A game I have long since tired of.
Is this story ringing any bells yet? The truth was that both articles quoted a paper which stated that 17.4% of people suffered adverse effects. So, yes, a pedant would say that the 18 – 20% figure was wrong – although not very wrong. Certainly not worth a demand of instant retraction, and apology, which is a very drastic step indeed.
Anyway, below is a short description of the findings of an independent panel set up by Fiona Godlee, editor of the BMJ, regarding the Rory Collins attacks:
“As previously reported, Rory Collins, a prominent researcher and head of the Cholesterol Treatment Trialists’ (CTT) Collaboration, had demanded that The BMJ retract two articles that were highly critical of statins. Although The BMJ issued a correction for both papers for inaccurately citing an earlier publication and therefore overstating the incidence of adverse effects of statins, this response did not satisfy Collins. He repeatedly demanded that the journal issue a full retraction of the articles, prompting The BMJ’s editor-in-chief, Fiona Godlee, to convene an outside panel of experts to review the problem.
The report of the independent statins review panel exonerates The BMJ from wrong doing and said the controversial articles should not be retracted:
“The panel were unanimous in their decision that the two papers do not meet any of the criteria for retraction. The error did not compromise the principal arguments being made in either of the papers. These arguments involve interpretations of available evidence and were deemed to be within the range of reasonable opinion among those who are debating the appropriate use of statins.”
In fact, the panel was critical of Collins for refusing to submit a published response to the articles:
“The panel noted with concern that despite the Editor’s repeated requests that Rory Collins should put his criticisms in writing as a rapid response, a letter to the editor or as a stand-alone article, all his submissions were clearly marked ‘Not for Publication’. The panel considered this unlikely to promote open scientific dialogue in the tradition of the BMJ.””1
To provide a bit more context at this point, you should know that for a number of years, people have been trying to get Rory Collins to release the data he and his unit (the CTT), holds on statins. [The CTT was set up purely to get hold of and review all the data on statins, it has no other function].
He has stubbornly refused to let anyone see anything. He claims he signed non-disclosure contracts with pharmaceutical companies who send him the data, so he cannot allow anyone else access. Please remember that some of the trials he holds data on were done over thirty years ago, and the drugs are long off patent. So how the hell could any data still be ‘confidential’ or ‘commercially sensitive’ now?
[The concept that vital data on drug adverse effects can be considered confidential, and no-one is allowed to see it, is completely ridiculous anyway. But that is an argument for another day.]
Now, amazingly, after running the CTT for nearly twenty years, Collins claims that ‘he has not seen the full data on side-effects.’ In an e-mail to the Sunday Express he stated that ‘his team had assessed the effects of statins on heart disease and cancer but not other side effects such as muscle pain.’
Let that statement percolate for a moment or two. Then try to make sense of it. So, they have got the data, but not bothered to look at it? Or they have not got it – which surely must be the case if he hasn’t even seen it. Give us a clue. Either way, Collins states he has not assessed it.
Despite this, he still managed a vicious attack on the BMJ for publishing articles, claiming statins had side effects of around 20%. This was an interesting stance to stake, as he now claims he has no idea what the rate of side effects are? In which case he should make a grovelling apology to Fiona Godlee immediately.
What is certain, and must be reiterated, is that Rory Collins has consistently refused to allow anyone to see the side effect data, or any other data, that that the CTT may, or may not, hold. See e-mail below from Professor Colin Baigent to the ABC producer MaryAnne Demasi (she was trying to get the CTT to confirm that they would not release data, Colin Baigent is, or was, deputy to Rory Collins)
From: colin.baigent@xxxxxxxxxxx
To: maryannedemasi@xxxxxxxxxxxx
Subject: RE: URGENT COMMENT NEEDED PLEASE: ABC TV AUSTRALIA
Date: Tue, 24 Sep 2013 17:02:23 +0000
Dear Maryanne
The CTT secretariat has agreement with the principal investigators of the trials and, in those instances where trial data were provided directly by the drug manufacturers, with the companies themselves, that individual trial data will not be released to third parties. Such an agreement was necessary in order that analyses of the totality of the available trial data could be conducted by the CTT Collaboration: without such an agreement the trial data could not have been brought together for systematic analysis. Such analysis has allowed the CTT Collaboration to conduct and report all of the analyses on efficacy and safety that have been sought directly or indirectly by others (eg by Dr Redberg in her papers on the efficacy and safety of statins in primary prevention, and in questions raised by the Cochrane Collaboration). Hence, the CTT Collaboration has made available findings that would not otherwise have emerged.
I would be very happy to ring you at whatever time is convenient for you in order to help you to understand our approach, and then address in writing any residual concerns. It would be a shame if we were not able to speak as this would be the most effective way of explaining things.
Please let me know where and some times when I can reach you, and I will endeavour to telephone.
Colin Baigent.
I put the word safety in bold in this copied e-mail. You will note that Professor Colin Baigent does not say that that the CTT do not have these data on safety. He just says that the CTT won’t let anyone else see any data.
If they do have it, why have they not done this critically important review before, as they have had much of the data for over twenty years. If they don’t have it, how exactly is Rory Collins going to review it – as he states he is going to? Sorry to keep repeating this point, but I think it is absolutely critical.
Picture the scene in a lovely oak panelled office in Oxford, the city of the dreaming spires….
Professor Collins: ‘Hey guys, you’re just not going to believe this, but a researcher just found a big box in the airing cupboard, and guess what, it has all the safety data in it….phew.’
Professor Baigent: ‘Ahem… Why that’s lucky Professor Collins, now we can do the safety review.’
Professor Collins: ‘Ahem… Indeed, Professor Baigent, we can. So, let’s get cracking shall we?’
And lo it has come to pass that after all these years Professor Collins has deigned to look at the safety data. This review shall, in Collins own words ‘be challenging.’ But you know what. I really don’t think they should bother, because we all know exactly what they are going to find….
That they were right all along, statins have no side effects. Hoorah, pip, pip. Nothing to see here, now move along.
A.N.Other Researcher: ‘Please sir, can anyone else see these data that you hold, to ensure that you are being completely open and honest?’
Professor Collins: ‘Don’t be ridiculous, these data are completely confidential.’
At this point I feel that I should ask how much do you, gentle readers, believe you can trust a review by Collins, on the data that Collins holds, on behalf of the pharmaceutical industry. Data that no-one else can ever see. [And the data from clinical trials on side effects is totally inadequate anyway].
Were I to be given the task of finding someone to review the safety data on statins, Professor Sir Rory Collins would not be the first person I would ask. He might even be the last.
P.S. Actually, he would be the last.
I do not claim to be Nostradamus here. What was going to happen was obvious. The script had been written a long time ago. It was only a question of when, not if, it happened.
However, whilst the article itself is nothing new… and believe me, there is nothing new here. Just the same data stretched into three hundred references, and mind-blowing statistical obfuscation. It does, however, contain a few new Alice in Wonderland statements, such as the following:
‘If information on a particular outcome is not available from a randomised trial because it was not recorded, that would not bias assessment of the effects of the treatment based on trials that did record that outcome.’ How can this statement be made? For the first twenty years of trials on statins, no-one had noted that statins increase the risk of type II diabetes. It was not, as far as could be seen at the time, a problem.
Then, in a later study, JUPITER, all of a sudden it was found that there was a significant increase in type II diabetes. Now, it turns out that all statins increase the risk of type II diabetes. Had JUPITER not recorded the incidence of type II diabetes, this would never have been noticed. The cynics among you might say that they recorded this in the hope that the incidence would actually go down.
Here we have a perfect example of an outcome not recorded in the vast majority of statin studies. Had it been, it would have significantly biased the assessment of treatment. We also find that after two trials, 4S and HPS, found an increase in non melanoma skin cancer2, this outcome was not recorded, ever again, in statin trials. Outcomes certainly cannot make a difference if you do not record them. But if you did bother record them – who knows what might have happened.
This type of logic litters this Lancet paper, along with straw man argument after straw man argument. However, the purpose of this blog was not to discuss the evidence, such as it is, such as we are allowed to see, but to highlight why this paper was written and published. For this I shall turn to the editorial, accompanying the paper, written by Richard Horton. Who is the editor of The Lancet.
Read this, and be afraid, for it is the most frightening thing you will read this year. Possibly this decade and maybe the entire century as is a direct attack on human freedoms. Whilst couched in the usual life destroying scientific prose, what he is saying is that any who questions current accepted medical dogma should be very tightly controlled, and probably should not be allowed to publish anything at all.
The entire editorial is an exercise in trying to silence any dissent with what some might view as threats and bullying. This, I think, is the key paragraph (my emphasis in bold).
‘The debate about statins, as for MMR, has important implications for journals. Some research papers are more high risk to public health than others. Those papers deserve extra vigilance. They should be subjected to rigorous and extensive challenge during peer review. The risk of publication should be explicitly discussed and evaluated. If publication is agreed, it should be managed with exquisite care.’
Now that, when you strip it down, is basically censorship.
Despite the seriousness of what Richard Horton is proposing, it is amusing to know what his published views on peer review might be, consider his statement that ‘Those papers deserve extra vigilance. They should be subjected to rigorous and extensive challenge during peer review’:
‘The mistake, of course, is to have thought that peer review was any more than a crude means of discovering the acceptability — not the validity — of a new finding. Editors and scientists alike insist on the pivotal importance of peer review. We portray peer review to the public as a quasi-sacred process that helps to make science our most objective truth teller. But we know that the system of peer review is biased, unjust, unaccountable, incomplete, easily fixed, often insulting, usually ignorant, occasionally foolish, and frequently wrong.’ https://en.wikipedia.org/wiki/Richard_Horton_(editor)
Anyway, you can read the editorial in full here (http://www.thelancet.com/pdfs/journals/lancet/PIIS0140-6736(16)31583-5.pdf). In addition to the paragraph highlighted above, I would like to draw your attention to a couple of other very worrying statements in the closing parapgraphs:
The Committee’s [Committee on Publication Ethics COPE] decision [not to investigate statin critics as demanded by ‘concerned’ scientists] points to a serious gap in UK science—the lack of a central institution where scientists who wish to question the actions or ethics of other scientists or scientific institutions can go. Allegations of research misconduct are best investigated by the institution where the original research took place. But that principle does not apply for some organisations, such as scientific or medical journals.
With no independent tribunal to consider allegations of research or publication malpractice, a damaging dispute has been allowed to continue unresolved for 2 years, causing measurable harm to public health.
The debate about statins, as for MMR, has important implications for journals. Some research papers are more high risk to public health than others. Those papers deserve extra vigilance. They should be subjected to rigorous and extensive challenge during peer review. The risk of publication should be explicitly discussed and evaluated. If publication is agreed, it should be managed with exquisite care.
Authors and editors should be aligned on the messages they wish to convey, and every eff ort must be made to avoid misinterpretations and misunderstandings in the media. Editors also have to separate their roles as gatekeepers and campaigners. It is tempting to publish science that confirms pre-existing beliefs, especially if those beliefs underpin a campaign. Two ongoing campaigns—against Too Much Medicine and for Statin Open Data—continue to imply that statins are overused and that hidden harms remain to be exposed. As the Review we publish makes clear, the best available evidence indicates that neither statement is true.
Would this be the same Richard Horton, editor of the Journal, the Lancet, who wrote? ‘Journals have devolved into information laundering operations for the pharmaceutical industry.’3
Would this be the Richard Horton who said? “The case against science is straightforward: much of the scientific literature, perhaps half, may simply be untrue. Afflicted by studies with small sample sizes, tiny effects, invalid exploratory analyses, and flagrant conflicts of interest, together with an obsession for pursuing fashionable trends of dubious importance, science has taken a turn towards darkness.”4
And would this be the same man who followed it up with?
‘The apparent endemicity of bad research behaviour is alarming. In their quest for telling a compelling story, scientists too often sculpt data to fit their preferred theory of the world. Or they retrofit hypotheses to fit their data. Journal editors deserve their fair share of criticism too. We aid and abet the worst behaviours. Our acquiescence to the impact factor fuels an unhealthy competition to win a place in a select few journals. Our love of “significance” pollutes the literature with many a statistical fairy-tale. We reject important confirmations. Journals are not the only miscreants. Universities are in a perpetual struggle for money and talent, endpoints that foster reductive metrics, such as high-impact publication.’4
Couldn’t have put it better myself. Yet, despite the fact that Richard Horton knows that much of the research is flawed and distorted by ‘flagrant conflicts of interest’ he still seems to believe that the statin studies, uniquely in history, are perfect – and cannot be questioned in any way. “Doublethink means the power of holding two contradictory beliefs in one’s mind simultaneously, and accepting both of them.” (George Orwell).
What do other editors think of this latest paper? Well, we have the thoughts of Fiona Godlee (editor of the BMJ), and Rita Redberg (editor of the Journal of the American Medical Association). I will supply a few quotes from them in an article published in Medpage Today (http://www.medpagetoday.com/cardiology/cardiobrief/60122):
‘More generally, Godlee and Redberg lamented the absence of independent verification of the statin data. Redberg said that “none of the CTT data has been made available to other researchers, despite multiple requests.” “No one has seen these data except the trialists.” Godlee agreed. “Ideally all clinical trial data should be available for third-party scrutiny,” she said.
Godlee’s also noted that “this is not an independent review, this is a review by the trialists.” Redberg went further, saying that “the long declaration of interests is telling. The Oxford Clinical Trials Unit receives hundreds of millions of pounds of support from the pharmaceutical industry.”
Godlee said that the need for independent review is especially pressing in this case, given the public health implications of the call for widespread use of statins for primary prevention. Redberg went even further and observed that “all of this data is from industry-sponsored studies, with concern for bias.”
As they went on to say
‘Redberg also pointed out some unintended consequences of statin usage. “Data shows that people on statins are more likely to become obese and more sedentary over time than non-statin users, likely because people mistakenly think they don’t need to eat a healthy diet and exercise as they can just take a pill to give them the same benefit (Sugiyama et al. JAMA IM 2014). So it seems this review affirms that many healthy people who feel perfectly well can take a pill every day, not live any longer, suffer any number of adverse effects, all to treat the ‘disease’ of LDL. I maintain the best way to reduce cardiac risk is to eat a Mediterranean-style diet, get regular physical activity, don’t smoke, and enjoy yourself.”
Godlee also emphasized the limitations of primary prevention. “Evidence about poor adherence to statins has long been known,” said Godlee. “People don’t want to take a drug forever. The problem didn’t arise with the BMJ study.”
It also seems likely that the Lancet paper exaggerated the benefits of primary prevention. The long-term benefits of primary prevention in the paper were based on modeling. The calculated benefits might have been a best-case scenario.’
In short, they did not think much of this paper, and Fiona Godlee was particularly concerned about the censorship element:
‘Godlee rejected the comparison of the BMJ papers to the Lancet Wakefield paper and objected to the idea that it’s too dangerous to publish papers critical of statins. “Where do you stop and where does that begin?” she wondered. She also pointed out that public concern over statins in the U.K. became elevated, not after the publication of the BMJ papers, but after Collins brought attention to the papers in a public denunciation of the papers on the BBC.
“We have to allow debate, I don’t know where you would draw the line,” she said. “In terms of public debate, the statin debate is fascinating and deserves airing.”’
So, thank goodness for them. I shall stop now, although there is much still to say, because this blog is already very long and people may fall asleep reading it. However, I think this is such an important issue – potential censorship in medical research – that I felt I absolutely had to write something. So, here it is.
I shall finish on two things. Firstly, to state the Uffe Ravnskov, who has been a long-term campaigner against the cholesterol hypothesis, and statins, had one of his books, burned, during a live television debate. I do not have any footage, but here is my attempt to replicate the scene using a photograph from the past.

Secondly, here is a list of some of the conflicts of interest of the authors of the paper.
Declaration of interests
JA, CB, LB, RC, JE, RP, DP, and CR work in the Clinical Trial Service Unit & Epidemiological Studies Unit (CTSU) at the University of Oxford. The CTSU has received research grants from Abbott, AstraZeneca, Bayer, GlaxoSmithKline, Merck, Novartis, Pfizer, Roche, Schering, and Solvay that are governed by University of Oxford contracts that protect its independence, and it has a staff policy of not taking personal payments from industry (with reimbursement sought only for the costs of travel and accommodation to attend scientific meetings). RC is co-inventor of a genetic test for statin-related myopathy risk, but receives no income from it. DP has participated in advisory meetings for Sanofi related to PCSK9 inhibitor therapy in his previous employment. The CTT Collaboration, which is coordinated by CTSU with colleagues from the University of Sydney, does not receive industry funding. JD has received research grants from, and served as a consultant to, Merck and Pfizer. GDS hast twice received travel and accommodation funding and honoraria from Merck; DD receives compensation for serving on data monitoring committees for clinical trials (including of statins) funded by Abbvie, Actelion, Amgen, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Merck, Sanofi , and Teva. NW and ML are inventors of a combination formulation for the prevention of cardiovascular disease that includes a statin, covered by patents licensed to Polypill in which they both hold shares and which owns the website polypill.com. SMac has received research grants for research on statins and polypill development from Bristol-Myers Squibb and BUPA. SMar is co-inventor on a pending patent for a LDL cholesterol estimation method, and has served as an advisor to Sanofi, Regeneron, Quest Diagnostics, Pressed Juicery, and Abbott Nutrition. NP has received research grants and honoraria for participating in advisory meetings and giving lectures from Amgen, Lilly, Menorini, and Merck. PR has received investigator-initiated research grants from Amgen, AstraZeneca, Kowa, Novartis, and Pfizer. PSa has received research grants and honoraria for consultancies from Amgen and Pfizer. LS has undertaken advisory work unrelated to statins for AstraZeneca and GlaxoSmithKline. SY has received a research grant from AstraZeneca through Hamilton Health Sciences. AR declares that George Health Enterprises, the social enterprise arm of The George Institute, has received investment to develop combination products containing statin, aspirin, and blood-pressure-lowering drugs. JS has received grants from the National Health and Medical Research Council, Australia; Bayer Pharmaceuticals; Roche; and Merck Serono. RB, SE, BN, IR, and PSa declare no competing interests.
[This list is far from complete. Paul Ridker, for example was (and may still be) a board member of Merck Sharp and Dohme, the maker of simvastatin at the time. Something he failed to report in a paper entitled: ‘Association of LDL Cholesterol, Non-HDL Cholesterol and Apolipoprotein B level with risk of cardiovascular events among patients treated with statins: A meta-analysis.’6 And something he has not mentioned here either.]
1: http://www.thelancet.com Published online September 8, 2016 http://dx.doi.org/10.1016/S0140-6736(16)31357-5
2: Hung SH, Lin SC, Chung SD. Statins use and thyroid cancer: a population based case-control study. Clin Enodocrinol (Oxf) 2014 published online 30 July 2014,doi:10.111/cen.12570
3: Richard Smith. “Medical journals are an extension of the marketing arm of pharmaceutical companies.” Public library of Science. (May 17, 2005).
4: http://www.thelancet.com Vol 385 April 11, 2015
5: http://www.medpagetoday.com/cardiology/cardiobrief/60122
6: Correction. “Unreported Financial Disclosures in: Association of LDL Cholesterol, Non-HDL Cholesterol, and Apoliprotein B levels with risk of cardiovascular events among patients treated with statins: a Meta-analysis.’ JAMA. (April 25, 2012].