In my book the Clot Thickens, and in other blogs and lectures, I have stated that LDL cannot get through the endothelium (lining of blood vessel walls) and into the artery wall behind. So, the idea of LDL leaking from the bloodstream and into the artery wall is nonsense.
Recently, however, people have been bombarding me with papers and AI generated essays, stating that there are clear mechanisms that allow this to happen. Indeed, it does happen – so they say. The primary pathway is transcytosis.
So, LDL can get into the arterial wall past the ‘barrier’ of endothelial cells, and the impenetrable tight junctions between them? Am I wrong about this?
What is transcytosis?
I probably need to begin by explaining what transcytosis is, in as few words as possible. Unfortunately, not that few. With some other background bits and pieces.
The first point I want to make is that all human cells ruthlessly control what is allowed into, and out of them. If they lose this control they will die, almost instantly. It is, pretty much the definition of cell death. This ‘substance control’ occurs all the way down to the atomic level – the smallest size possible.
By which I mean that even ions (charged atoms) such as sodium, chloride, potassium and calcium have to pass through gates, or channels, to be allowed entry or exit. This is a tightly controlled process, requiring cellular energy, and a large number of complex mechanisms, including messenger molecules. The gates/channels are embedded in the cell membrane.
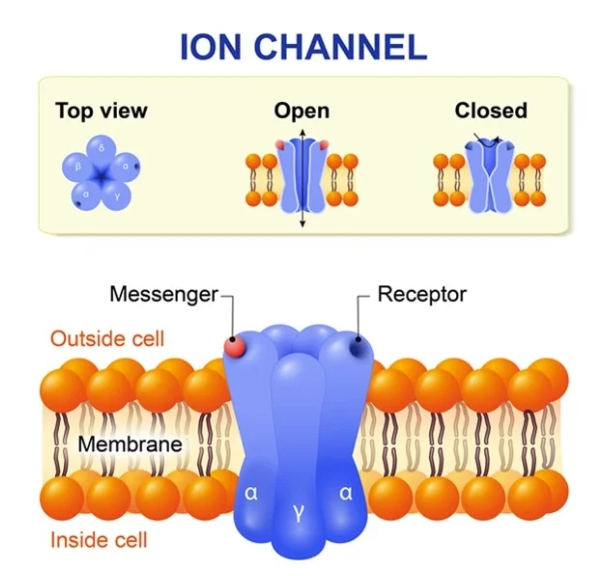
LDL molecules are thousands of times bigger than an atom. Ergo, there is simply no way they can force entry into a cell. [Terms and conditions apply, see under viruses].
Given this, how does LDL get from the bloodstream into endothelial cells – which it clearly does? The answer is that the cell manufactures a receptor, the LDL receptor which then travels to the cell membrane – where it embeds itself, ready to grab a passing LDL. This system was first identified by Goldstein and Brown – who won the Nobel Prize for their work in this area.
How does the receptor operate? Very simply, LDL has a protein attached to the side of it, the ApoB-100 protein, which is the ‘key.’ This key is perfectly designed to fit into the LDL receptor ‘lock’. Once locked on, the LDL molecule and the receptor are then pulled into the cell through a process known as endocytosis.
In essence the cell membrane wraps round the LDL/receptor complex forming a little lipoprotein sphere. Once inside the cell, the sphere is broken apart releasing its contents – including cholesterol and fat(s). And a few other things.
The next step is that the LDL receptor itself is – often but not always – broken down by an enzyme called, – wait for it – Proprotein convertase subtilisin/kexin type 9 (PCSK9). If the receptor is not disintegrated by this enzyme, it recycles back to the cell membrane, ready to lock onto another LDL molecule.
Most cells have thousands of LDL receptors stuck to the side at any one time. So, this is not a cottage industry, it is heavy-duty manufacturing.
That is how LDL can get into a cell. Through ‘endocytosis.’ ‘Endocytosis is the process by which cells absorb substances by engulfing them.’
[Endocytosis is also, mostly, how viruses get into cells. Viruses have proteins attached to their outer casing, such as the spike protein on Sars-Cov2. If this spike can find something to lock onto – in this case the A2 receptor – it will be ‘endocytosed’ and pulled into the cell. However, if there is no lock to be found, the virus cannot enter, and you cannot be infected by that virus. Interestingly viral particles and LDL molecules are pretty much the same size].
Below is the original diagram of the LDL receptor. It does not include PCSK9, because no-one knew it existed at the time.
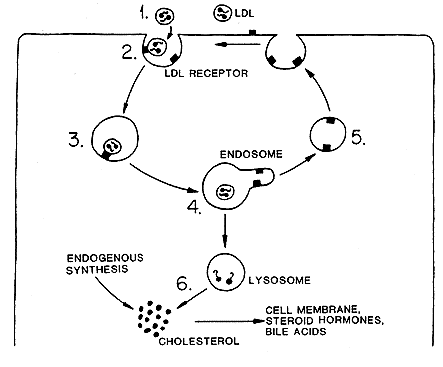
All of this is very widely accepted as fact – even by me.
So, there is no issue with getting LDL into a cell, which is the first step in transcytosis. Can LDL then be transported across the cell and popped out the back, into the arterial wall behind, a process called ‘exocytosis’? The full mechanism of transcytosis has three parts? Entry, then transport across the cell, then exit. Endo-trans–exo .. cytosis.
Problems
The first point I would like to make here is one of scale. If you were the size of an LDL molecule, the cell would be two kilometres across – approximately. Therefore, LDL is not going to simply drift from one side to the other, then bump into the cell membrane on the other side. Well, it might, but it would take a hell of a long time to do so.
Instead, it needs to be actively transported through the cell – in some way. Which means that, for transcytosis to work, the cell needs to have complex mechanisms within it designed to take LDL by the hand and lead it through the cell. Then exocytose it out the back. This is not some random – a million chimps writing Shakespeare – type of thing.
In super-simplified diagrammatic version, it would look something like this:
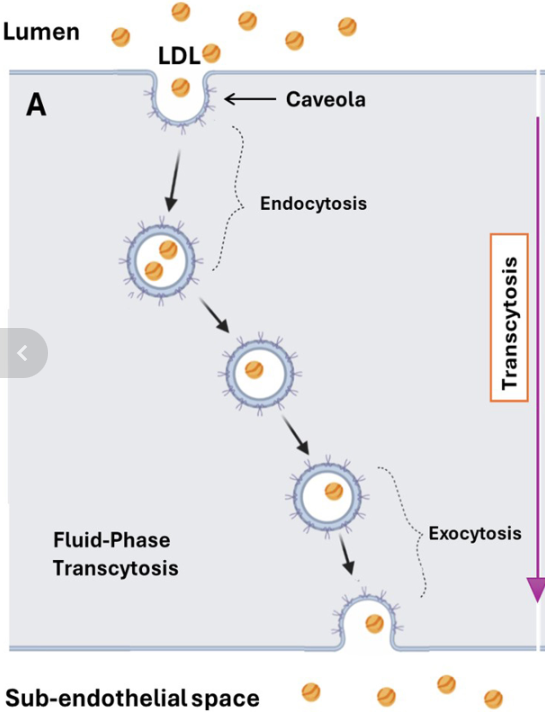
It is true that cells do possess mechanisms that enable them to transport various molecules from one side to the other. Then pop them out the back. Although, with LDL, the ‘how’ remains unclear, and vague. And the ‘why’ is even more uncertain.
As in, why would an endothelial cell go to all the trouble of absorbing an LDL molecule – then decide not to break it down – then transport it across itself before popping it into the sub-endothelial space behind? [A very narrow fluid filled space]
The only reason for this must be that the cells behind, in the deeper layers of the arterial wall, need cholesterol, and require it to be passed on to them. How would they communicate this need to the endothelial cell?
Well, for this to happen the cells in the arterial wall would need to synthesize a messenger molecule that travels through their cell membrane – then crosses the subendothelial space to lock onto a receptor on the outside of the endothelial cell membrane facing the arterial wall.
This would trigger another messenger molecule to say that the cells behind need LDL. So, could you please not break down all the LDL molecules you absorb and send them through to us instead. Yes, indeed, it’s a bit of a long and complicated message, but it could happen. The body acts in mysterious ways, its wonders to perform. Or something like that.
You may choose to believe that all such mechanisms, and messenger molecules, and the receptors for said molecules to lock onto exist, and have been fully identified … and have been seen operating in vivo. Or you may not.
But you can hopefully understand that we are not talking about some passive, ‘LDL moving down a concentration gradient from blood to arterial wall thing’ here. Sailing happily from the bloodstream directly into the arterial wall. You may also see why I am highly sceptical about all of this. Primarily, because it does not make any sense at all [see under vasa vasorum, discussed later].
Why so much recent focus on transcytosis?
Transcytosis has become an area of much research, and comment, recently. Why? Well, I like to think I may have been the cause of some of it. For I have stated in books, lectures, and writing that LDL cannot simply slip through – or past – the endothelial barrier. Thus, LDL cannot passively leak into the arterial wall, causing plaques. Ergo, the LDL hypothesis is wrong.
My position on this is, I believe, based on rock-solid logic. If LDL cannot navigate a way through undamaged endothelial cells, and the tight junctions that bind endothelial such cells together, then LDL cannot be the proximate cause of plaque development. Because it cannot get into the arterial wall to start creating a plaque in the first place.
I think this has, finally, been recognized as being a rather major obstacle to the LDL hypothesis. Whether through my work and writings, or for other reasons. Whichever, it now seems to have become more widely accepted that the concept of LDL travelling down a concentration gradient is scientific nonsense. And always has been. Sigh … head hits desk
Just to start with, a concentration gradient simply cannot shift things out of the bloodstream and into any cell. Because, sitting between the blood, and any cell, is a cell membrane. And, apart from anything else, you cannot have a concentration gradient between two ‘fluids’ if there is a solid barrier lying between them. No ‘gradient’ exists unless the barrier breaks down. Then things can travel about.
More relevant is the fact that cells are perfectly capable of shifting molecules up – against any ‘theoretical’ concentration gradient. They do this all day, every day. In fact, they do this millions of times a day. If this didn’t happen, we would cease to exist. Your heart would instantly stop beating, for example.
Here, from Wikipedia, is a short explanation of the sodium-potassium pump – which pumps sodium out of cells, and potassium in:
‘The sodium–potassium pump also known as Na+/K+-ATPase, Na+/K+ pump, or sodium–potassium ATPase) is a found in the cell membrane of all animal cells. It performs several functions in cell physiology.
The Na+/K+-ATPase enzyme is active (i.e. it uses energy from ATP) For every ATP molecule that the pump uses, three sodium ions are exported and two potassium ions are imported. Thus, there is a net export of a single positive charge per pump cycle. The net effect is an extracellular (outside the cell) concentration of sodium ions which is 5 times the intracellular (inside the cell) concentration, and an intracellular concentration of potassium ions which is 30 times the extracellular concentration.’
Yes, cells can pump potassium ions out and into the blood, or elsewhere, ‘against’ a massive concentration differential. And the potassium ions do not simply leak back in again, because they can’t. Because the cell membrane acts as a barrier to re-entry.
And just to boggle your mind a bit more there can be, up to, thirty million sodium-potassium pumps on an individual cell. [Nerve cells have the most, it’s how they pass electrical messages].
And an average endothelial cell will have around forty thousand LDL receptors on its surface. Which may give you some idea of how vital cholesterol is for healthy cellular function.
The point I am trying to emphasize here, perhaps over-emphasize, is that hundreds of different molecules move in and out of cells, under the control of complex cellular processes, requiring receptors and messenger molecules Nothing happens here that is not tightly controlled. And I mean nothing – viruses aside.
For many years there has been a horribly lazy assumption that, if the LDL concentration in the blood is high, it will simply move out of the bloodstream into the artery wall. Straight though the endothelial barrier.
Rather belatedly it has become more widely accepted that for the LDL hypothesis to work, there must complex mechanisms in place to allow LDL to travel though endothelial cells and into the ‘sub-endothelial’ space behind. And then get taken up into the arterial wall – in some mysterious way.
And lo, it seems, we now find that these mechanisms exist. Or do they? Small parts of the mechanism have been proven to exist, in some cells. As for the whole linked together system … nope. Not even close.
Some real-world stuff – PCSK9 Inhibitors and the vasa vasorum
PCSK9 Inhibitors
I hope to have made clear there are many (theoretical) areas where the transcytosis conjecture falls to bits. What about in the real world? For now, I am only going to look at two issues. I may look at others a bit later.
The newest LDL lowering drugs to hit the market are PCSK9 inhibitors. The thinking behind them is that, if you can block PCSK9, then fewer LDL receptors will be broken down. Therefore, more receptors will travel back to the cell membrane, more LDL will be absorbed by cells, and the LDL level will fall – thus reducing the risk of cardiovascular disease.
These wonder drugs include:
- Evolucamab (Repatha)
- Alirocumab (Praluent)
- Inclisiran (Leqvio)
It is true that they can reduce LDL in the bloodstream by, well over, fifty per cent. A much greater reduction than can be achieved with statins. But you may have just spotted a problem with the logic.
If more LDL is taken up by endothelial cells, then (if the transcytosis argument is correct) more ‘non-broken-down LDL’ will be available to be transported into the arterial wall behind – to form plaques. Ergo, you will be increasing the risk of cardiovascular disease, not reducing it.
So, do they work, or not? Well, I have looked at the studies and whilst they do not significantly increase the risk of cardiovascular disease, they have spectacularly failed to show much benefit. Here from a review of the FOURIER trial.
‘After readjudication, deaths of cardiac origin were numerically higher in the evolocumab group than in the placebo group in the FOURIER trial, suggesting possible cardiac harm.’ 1
Yes, reducing PCKS9 levels is amazingly effective at lowering of LDL, whilst slightly increased risk of cardiovascular death. Transcytosis argument proven, or disproved? I think disproved.
Perhaps more important, if not directly related to this discussion, is that they just managed to spectacularly disprove the LDL hypothesis. Sixty per cent lower LDL leading to an increased rate of cardiovascular deaths. Moving on.
The vasa vasorum
All arteries, of the size where plaques develop, have their own blood vessels lying within called vasa vasorum (blood vessels of the blood vessels). They supply nutrients to the arterial wall. It is their role in life.
Or, to look at this in another way, the cells in the arterial wall do not require access to the nutrients in the bloodstream that flow through the artery. If they need cholesterol from LDL molecules, for example, they can get it. Because it is travelling all around them in very small blood vessels which are purely designed to provide all the things the cells within arteries need.
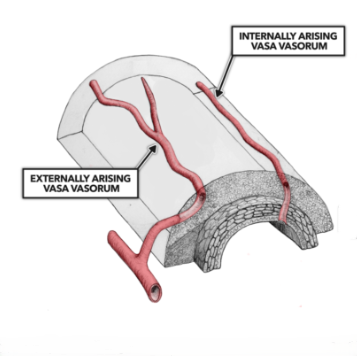
Yes, these vasa vasorum are lined with endothelial cells – as are all blood vessels. However, at the smallest ‘capillary’ size blood vessels the endothelium does not act as a barrier. They are, usually, leaky, and made deliberately so. Because, once blood arrives at its final destination substances have to be able move in and out of the blood freely.
Blood arriving at the kidneys, for example, has to unload waste products into each nephron – for ejection in the urine. Or the kidneys won’t work. And blood vessels in the liver are equally leaky.
In fact, just to complicate things further, there are three different versions of capillaries [smallest blood vessels in the body]..
Continuous, where the endothelial cells are tightly bound together – and wrap back onto themselves. In addition, the basement membrane supporting them is also continuous. Continuous capillaries retain control of the passage of all molecules. They are found, mainly, in the brain, and this structure creates the blood brain barrier.
Then there are fenestrated capillaries, with small holes in the endothelium.
Finally, there are sinusoidal capillaries. These are very leaky, and are found within the vasa vasorum.
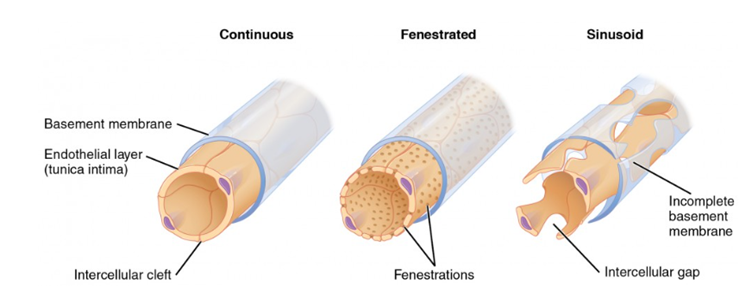
At this point, to my mind, the entire transcytosis argument becomes pretty much irrelevant. Why? Because LDL can get into the artery wall any time it likes, via the vasa vasorum. There is no barrier here. Entry by the back door, if you like.
So why have we become tangled up with complex arguments on transcytosis? If f there is a simple way that LDL can enter the artery wall any time it likes?
Because gentle reader, veins have more vasa vasorum than arteries. So, LDL can enter vein walls with even greater ease than artery walls. In addition the blood vessels in the lungs (pulmonary vessels) also have vasa vasorum.
And yet …
Plaques do not (except in exceptional circumstances) develop in veins or pulmonary blood vessels. I would say never, ever, but it can happen with, for example, Eisenmenger’s syndrome. [Very high blood pressure in the lungs].
All of this leaves the LDL hypothesis with a logical chasm to bridge. Starting with the first issue:
Why would LDL transcytosing into artery walls cause atherosclerotic plaques to develop, when LDL transcytosing into vein walls, and pulmonary arteries, does not?
Just in case you are wondering, the LDL level in veins is significantly higher than in arteries:
‘The present study’s main finding is that all lipoproteins are found in lower amounts in aortic blood when compared with peripheral venous blood’ 2
The ultimate ad-hoc hypothesis
I may seem to have come an awful long way to simply wrap back round on myself, and dismiss the entire concept. But transcytosis is presented in such a ‘clever’ way that it sounds entirely plausible. It is also made to sound difficult, and virtually incomprehensible. How can you argue against something that you do not understand? The fear of sounding stupid looms large.
The reality is that transcytosis sprang to life as an ad-hoc hypothesis. Here I quote AI on the matter of such hypotheses:
An ad hoc hypothesis is a provisional explanation or supplementary assumption added to a theory specifically to save it from being disproven or falsified by new, conflicting evidence.
Key Characteristics
- Crisis-Driven: It is invented to explain an anomaly that the original theory failed to anticipate.
- Lacks Independent Testability: It usually cannot be tested or verified on its own; its only evidence is the problem it attempts to excuse.
- No Predictive Value: It does not lead to new discoveries or broader scientific understanding
Transcytosis may seem, superficially, to provide an answer to the question ‘how can LDL move from the bloodstream into the artery wall – though an impenetrable barrier, and thus cause plaques.’ It happens by transcytosis. Phew, the LDL hypothesis is still correct.
But:
- LDL can get into the artery wall via vasa vasorum
- Thus, it can also get into vein walls and pulmonary vessel walls in the same way
- So, why does LDL not cause plaques in these blood vessels?
Which leads us back, as always, to the same, critical question. What is different about arteries in the systemic circulation* that makes them prone to develop plaques, when the same level of LDL has no such effect on, or in, any other vessels?
*The systemic circulation means blood leaving the left ventricle, before travelling round the body and arriving back at the right atrium. From where it is pumped into the right ventricle and then goes into the lungs, before arriving back at the right atrium – the pulmonary circulation.
As I hope you can see, invoking transcytosis to explain anything gets you nowhere, as all the blood vessels, large enough to develop plaques, have vasa vasorum in them that allow free passage of LDL.
Next ad-hoc hypothesis will be, I guess: “Why transcytosis only happens in arteries – at the exact point where plaques develop”.
Anyway, I do hope you have learnt something new. You may wish to explore what I have written in more detail. You may want to attack my reasoning. I welcome that. One of the main problems in science nowadays is that no-one debates anything.