A few weeks ago, a sixth year student at Westminster School sent me an essay she had written on cholesterol, and why it does not cause CHD. She wants to go to medical school. No one made her do any of this. She just looked at the evidence and made her mind up. She wrote me this e-mail
Dear Dr Kendrick,
I am a final year student at Westminster School who intends to study medicine. I am extremely interested in your research and reading your book enthused my and led me to spend a large portion of my time researching studies which you and other authors on the same topic have referenced. The Chief Medical officer came to speak to us today and after her talk I quizzed her about what I have read in your book as well as a large wealth of research I have done myself. (Attached, if you care to look, is a copy of an essay I wrote which won the top prize in school essay competition based on this research). She was extremely defensive of the cholesterol causes heart disease hypothesis and claimed that NICE had on a population level declared this to be the case. She said that the evidence did not add up on a small study level, but when studies were put together (I assume by NICE) that the conclusion is in favour of cholesterol causing CHD.
I would love to know your thoughts on this and where I can find this population based evidence.
Kind regards,
Francesca Greenstreet
I wrote back to her, to say that there was no population based evidence. Or, if there was, it very clearly demonstrated no link between cholesterol levels and heart disease. The Chief Medical Officer was just blustering – as most people do when confronted with someone who dares to question medical dogma.
I thought her essay was extremely well written and makes all the points that I have been making for years. It is just gratifying to see that the evidence on cholesterol and heart disease is clear to anyone with a brain.
In Defence of Cholesterol
The American government, the British government and the NHS, three venerable bodies respected as sources of dietary advice, currently recommend a diet low in saturated fat and cholesterol.[1] The predominant reason this advice is given is the accepted belief held within the scientific community that high serum cholesterol levels are linked causally with the accumulation and build up of atheromas which lead to atherosclerosis and Coronary Heart Disease (CHD).
The commonly accepted and taught theory which links cholesterol to heart disease, the Lipid Hypothesis, states that cholesterol is carried from the liver to the rest of the body’s cells in Low Density Lipoproteins (LDLs) and carried back from the rest of the body’s cells to the liver in High Density Lipoproteins (HDLs). After being transported back to the liver by HDLs, Cholesterol is broken down by the liver or passes out of the body as a waste product. The Lipid Hypothesis states that eating saturated fat raises LDL levels. The cholesterol from LDLs forms fatty deposits, atheromas, which build up beneath the endothelium of the arteries. The build up of atheromas narrows the arteries and pieces of the atheromas can break off and become lodged in narrower arteries .Clots can form in the narrowed arteries which prevent blood flow and can starve organs of oxygen and nutrients. When clots or blockages form in the coronary arteries, necrosis occurs. This leaves part of the heart muscle not contracting and relaxing and can lead to a myocardial infarction.[2]
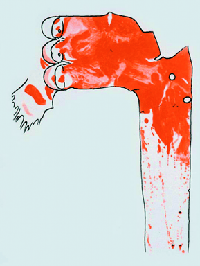
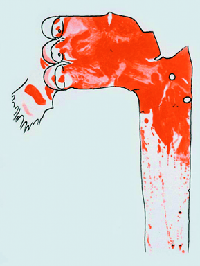
Figure 1: Sudan-stained aorta of a rabbit fed 61 egg yolks over a 70-day period, showing lesions in red.
The Lipid Hypothesis was brought to attention following a series of studies, the first of which was carried out by Anitschkow, a Russian scientist, in 1913. Anitschkow fed rabbits a diet of purified cholesterol dissolved in sunflower oil and examined the cells and the arteries of the rabbits after killing them. [3] Rabbits which were fed the purified cholesterol were found to have vascular lesions which bore a close resemblance to atheromas found in humans (Figure 1). Following Anitschkow’s study, Dr John Gofman led a team to similar findings and hypothesized that serum cholesterol was the cause of the lesions developing.[4] The similarity of the lesions to those found in humans suffering from CHD was catalytic in the formation of theories that a high cholesterol diet might be linked to CHD in humans.
The ideas behind the Lipid Hypothesis were formalised by Ancel Keys when, in a study in 1953, he used data from six countries to show a direct link between the percentage calories from fat in the average diet and the number of CHD deaths per 1000.[5] Furthermore, he found the incidence of CHD deaths in those six countries was best predicted by the intake of saturated fat.[6]
However, not all scientists and physicians are in agreement with the Lipid Hypothesis. Regarding Anitschkow’s rabbit study, it has been pointed out that cholesterol does not form part of the natural diet of a rabbit and thus it is possible that the rabbits had an allergic reaction to the high cholesterol diet, or that they were otherwise incapable of processing the chemical. It is significant to note that similar experiments carried out on dogs and rats showed that a rise in blood cholesterol did not lead to a rise in atherosclerosis.[7] This is potentially due to the fact that dogs and rats, unlike rabbits, consume cholesterol as part of their natural diet. The lack of cholesterol in a rabbit’s natural diet, combined with the failure to replicate the findings in dogs or rats, whose natural diets are much more similar to our own, is a significant flaw in the reasoning behind Anitschkow and Gofman’s conclusion: that a high cholesterol diet is linked to atherosclerosis in humans.
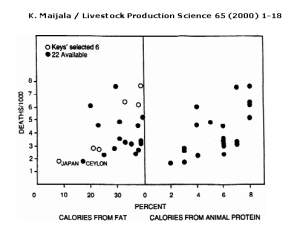
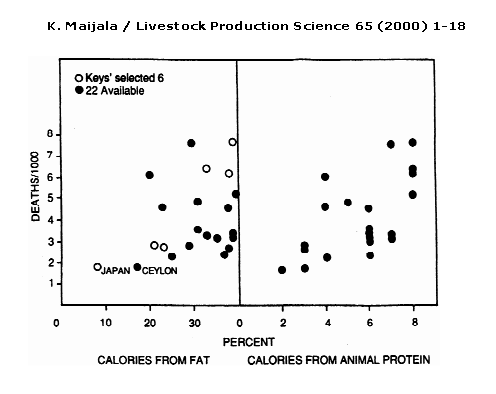
Figure 2: Keys’ (1953) selection to show relationship of fat intake to heart disease deaths of 55–59 yr. old men in 1951–53 (open circles left)and the 15 other available countries (closed circles). The relation of heart diseases to animal protein intake is on the right (Mann, 1993). (Adapted from WHO Ann. Epid. and Vital Statistics).
Another flaw in the Lipid Hypothesis is that Ancel Keys selected with purpose the countries for which he presented data in his study in 1953, rather than choosing them at random. “Yerushalmy and Hilleboe (1957) observed that Keys would have had available data from 22 countries, which would have given a much weaker correlation “(Figure 2).[8]
Figure 2 shows a very weak correlation between deaths per 1000 (from CHD) and percentage of calories from fat when all 22 countries are plotted on the same graph. It is interesting to note that the correlation between deaths per 1000 (from CHD) and percentage of calories from animal protein has a similar and even slightly stronger correlation than between deaths per 1000 (from CHD) and percentage of calories from fat. All of this data would have been available to Keys, so his focus on the link between percentage of calories from fat and the number of deaths per 1000 (from CHD) is curious.
The data sample presented by Keys gives a correlation of coefficient of +0.84, a strong positive correlation, whereas “in the simulation study by Wood (1981) on the consumption statistics of 21 countries a total of 116280 different samples of six countries were found, and the correlation between consumption of animal fat and CHD mortality varied from -0.9 to +0.9, the average being -0.04.”[9] Such a difference in correlation coefficients between similar studies indicates some bias in Keys’ selection of the six countries or insufficient data, since Wood’s study uses many more countries and therefore is more likely to be accurate. As it is obvious that Keys had sufficient access to data from the 22 countries, it seems that his selection was biased.
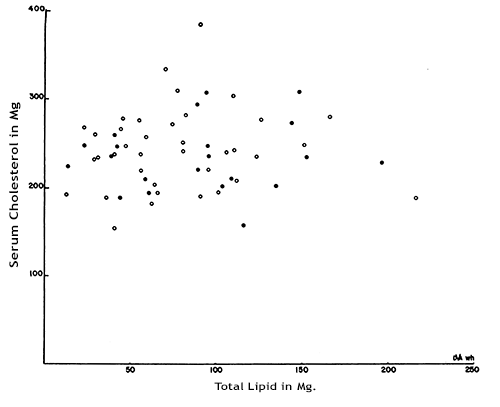
There have also been many studies which investigate serum cholesterol level in relation to atherosclerosis and CHD in Humans rather than in animals. A notable example is the study led by Paterson entitled: “Serum Cholesterol Levels in Human Atherosclerosis”. 800 patients who were permenantly confined to hospitals and 100 war veterans who were in hospital for dimiciliary care were given 2500-3000 calories a day in their daily diet, of which 25 to 35% was derived from fat. Serum cholesterol was determined annually or semi-annually and when any patients died, the severity of atherosclerosis was determined using six differnet criteria: crude morphological grading, measurement of the thickness of the largest plaque, determinations of the total lipid content, lipid concentration, total calcium content and calcium concentration. Figure 3 shows a graph showing the abscence of a correlation between Serum Cholesterol in mg.% and Total Lipid in Mg. This shows that for the criterium of total lipid content, there is no correlation in the age group of 60-69 years.
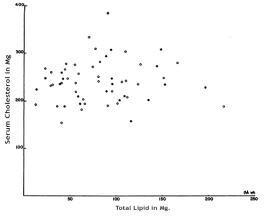
Figure 3: Total coronary artery lipid and serum cholesterol levels in patients 60-69 years. The open circles represent cases without complications of coronary atherosclerosis; the closed circles, cases with complications.
Similar findings were observed in the other age groups with a significant number of participants (70-79 and 80-89). The study concludes: “In the 58 cases in the age group 60-69 years, significant relationships between the serum cholesterol and the severity of the disease were found only once in 40 statistical analyses, and the complications of atherosclerosis were just as frequent in cases with low serum cholesterol levels (150-199 mg. %) as in cases with moderately high ones (250-299 mg. %).”[10]Considering the emphasis from the government and the NHS to reduce cholesterol and saturated fat intake because cholesterol causes heart attacks, this seems to be a remarkably weak correlation.
The Paterson study was not alone in its findings: Sigurd Nitter-Hauge and Ivar Enge published a study in The British Heart Journal in 1973 which reported: “No significant correlation was found when total coronary arterial score was correlated to serum cholesterol values or to triglycerides.”[11]
Not only is there strong evidence to show that serum cholesterol levels have no link to atherosclerosis, but there is also strong evidence to suggest that high cholesterol consumption does not raise blood cholesterol levels. The Framingham Heart Study, which set out to prove that eating more cholesterol in your diet increases your blood cholesterol levels, in fact showed that there was minimal difference in the blood cholesterol levels of the subjects despite subjects consuming cholesterol in widely varying amounts. [12] Scientists working on the Framingham Heart Study also studied the intake of saturated fats but eventually concluded: “There is, in short, no suggestion of any relation between diet and the subsequent development of CHD in the study group.”[13] It is difficult to stress the importance of this finding enough: there was no connection found whatsoever between diet and the development of CHD.
Further evidence that eating a diet high in saturated fat does not lead to CHD was published in The American Journal of Clinical Nutrition in 1981. The study compared the diets of two populations of Polynesians living on atolls near the equator. It also assesses the effect the diets have on the serum cholesterol levels in the populations. One of the populations, the Tokelauans, obtained a very high percentage of energy from coconut (high in saturated fat) compared to the Pukapukans, 63% compared with 34%. The Tokelauans had serum cholesterol levels 35-40mg higher than the Pukapukans. However , “vascular disease is uncommon in both populations and there is no evidence of the high saturated fat intake having a harmful effect in these populations.”[14]
Taking all these studies into account, it would appear that not only does having a high serum cholesterol level not have any connection to CHD, but that a diet high in cholesterol does not lead to high blood cholesterol levels and that a diet high in saturated fat does not have any link to CHD.
One final argument used to support the Lipid Hypothesis is the apparent effectiveness of statins in treating CHD. If, so the argument goes, statins reduce levels of serum cholesterol and they also reduce the risk of CHD, then reducing serum cholesterol levels must be the reason for the lower incidence of CHD. However, this reasoning contains two fallacies: firstly it assumes that statins have been shown to reduce the risk of CHD, and secondly it assumes that lowering serum cholesterol levels is the only effect of statins that could lower the incidence of CHD.
Both of these assumptions are false. A study was carried out by the University of British Columbia, part of the not-for-profit Cochrane collaboration, which concluded: “If cardiovascular serious adverse effects are viewd in isolation, 71 primary prevention patients have to be treated with a statin for 3 to 5 years to prevent one myocardial infarction or stroke.”[15] Small mortality benefits from statins have been shown for high-risk middle aged men.[16], [17] However, these trends are not seen in women and the elderly.15, 16 Even an advertisement for LIPITOR (atorvastatin calcium), one of the best-selling statins in America, has a disclaimer which includes: “LIPTOR has not been shown to prevent heart disease or heart attacks.”[18]
For the small minority of people who are protected by statins, there is another explanation. Statins have been repeatedly shown to act as “potent anti-inflammatory” agents in patients with cardio vascular disease[19]. The reason for their effects in reducing incidence of CHD could be due to those effects rather than the reduction of serum cholesterol levels. This means that it is innappropriate to use the limited protection against CHD by statins as evidence for high serum cholesterol levels being a cause of CHD.
Gathering together the arguments made in this essay, we can conclude that it is very likely that there is absolutely no causal correlation between high cholesterol, either in the serum or in the diet, and CHD. Nor is there any causal correlation between a diet high in saturated fat and CHD. The emphasis placed on the Lipid Hypothesis by the government and other organisations concerned with public health is potentially due to the inital panic after the publication of Keys’ and Anitschkow’s studies. The feeling of urgency to act in order to prevent ever increasing numbers of deaths from CHD led to premature acceptance of the Lipid Hypothesis without sufficient evidence. The long term effect of this view has been the demonisation of diets high in saturated fat and cholesterol without sufficient justification from otherwise reputable organisations for the past thirty years.
Word count : 2431 words
(excluding title, name, biliograph and references)
Bibliography
“Trick and Treat” (Barry Grroves,2008)
“The Great Cholesterol Con” (Dr Malcolm Kendrick, 2007)
“The Cholesterol Myths” (Uffe Ravnskov, M.D., Ph.D., 2000)
“Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease” (Ancel Keys, 1980, Introduction, p. 1-17)
KIM CRAMER, SVEN PAULIN, LARS WORKÖ, 1966. Coronary Angiographic Findings in Correlation with Age, Body Weight, Blood Pressure, Serum Lipids, and Smoking Habits. Circulation; 33:888-900
U. RAVNSKOV, 2002. Is Atherosclerosis caused by high cholesterol?. Q J Med; 95:397-403
Prevention of Coronary Heart Disease. British Medical Journal , 21 September 1968; No. 5620, p.689
Sigurd Nitter-Hauge, Ivar Enge, 1973. Relation between blood lipid levels and angiographically evaluated obstructions in coronary arteries British Heart Journal. 35, 791-795.
J. R. CROUSE, J. F. TOOLE, W. M. MCKINNEY, M. B. DIGNAN, G. HOWARD, F. R. KAHL, M. R. MCMAHN, G. H. HARPOLD, 1987. Risk factors for extracranial carotid atherosclerosis. Stroke; 18:990-996
Mukesh K. Jain, Paul M. Ridker, 2005. Anti-Inflammatory Effects of Statins: Clinical Evidence and Basic Mechanisms, Nature Reviews Drug Discovery 4. 977-987
Ian A. Prior, M.D., F.R.C.P., F.R.A.C.P., Flora Davidson, B.H.Sc., Clare E. Salmond, M.Sc., and Z. Czochanska, DIP.AG., 1981. Cholesterol, coconuts, and diet on Polynesian atolls: a natural experiment: the Pukapuka and Tokelau Island studies. The American Journal of Clinical Nutrition 34, p. 1552-1561.
Kannel WB, Gordon T., 1970.The Framingham Diet Study: diet and the regulations of serum cholesterol (Sect 24). Washington DC, Dept of Health, Education and Welfare.
J.C. PATERSON, M.D., LUCY DYER, M.Sc. and E.C. ARMSTRONG, M.D., London, Ont., 1960. Serum Cholesterol Levels in Human Atherosclerosis. Canad. M. A. J., 1960, vol. 82
Scandinavian Simvastatin Survival Study Group, 1994. Randomised trial of cholesterol lowering in 4444 patients with CHD: the Scandinavian Simvastatin Survival Study (4S). Lancet; 344: 1383-1389
Kalle Maijala, 2000. Cow milk and human development and well-being. Livestock Production Science 65 1–18
Daniel Steinberg, 2004. Review series: The Pathogenesis of Atherosclerosis. An interpretive history of the cholesterol controversy: part I, The Journal of Lipid Research, 45, 1583-1593.
GOFMAN, J.W.;LINDGREN, F.; ELLIOT, H.; MANTZ, W.; HEWITT, J.; STRISOWER,B.; HERRING, V.; LYON, T.P., 1950.The role of lipids and lipoproteins in atherosclerosis, American Association for the Advancement of Science Vol. 111pp. 166-171; 186
National Service Framework for Coronary Heart Disease – Modern Standards and Service Models, March 2000.
http://www.nhs.uk/conditions/cholesterol/pages/introduction.aspx
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001224/
http://www.health.gov/dietaryguidelines/dga2000/document/choose.htm
Figure 1: Daniel Steinberg, 2004. Review series: The Pathogenesis of Atherosclerosis. An interpretive history of the cholesterol controversy: part I, The Journal of Lipid Research, 45, 1583-1593.
Figure 2: Kalle Maijala, Cow milk and human development and well-being, Livestock Production Science 65 (2000) 1–18
Figure 3: J.C. PATERSON, M.D., LUCY DYER, M.Sc. and E.C. ARMSTRONG, M.D., London, Ont., 1960. Serum Cholesterol Levels in Human Atherosclerosis. Canad. M. A. J., 1960, vol. 82
[1] http://www.health.gov/dietaryguidelines/dga2000/document/choose.htm
[2] http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001224/
[3] Daniel Steinberg, 2004. Review series: The Pathogenesis of Atherosclerosis. An interpretive history of the cholesterol controversy: part I, The Journal of Lipid Research, 45, 1583-1593.
[4] GOFMAN, J.W.;LINDGREN, F.; ELLIOT, H.; MANTZ, W.; HEWITT, J.; STRISOWER,B.; HERRING, V.; LYON, T.P., 1950.The role of lipids and lipoproteins in atherosclerosis, American Association for the Advancement of Science Vol. 111pp. 166-171; 186
[5] “Trick and Treat” (Barry Groves, 2008, p.61-62)
[6] “The Cholesterol Myths” (Uffe Ravnskov, M.D., Ph.D., 2000, out of print – available at: http://www.ravnskov.nu/myth4.htm)
[7] “Trick and Treat” (Barry Groves, 2008, p.59)
[8] Kalle Maijala, Cow milk and human development and well-being, Livestock Production Science 65 (2000) 1–18
[9] Kalle Maijala, 2000. Cow milk and human development and well-being. Livestock Production Science 65 1–18
[10] J.C. PATERSON, M.D., LUCY DYER, M.Sc. and E.C. ARMSTRONG, M.D., London, Ont., 1960. Serum Cholesterol Levels in Human Atherosclerosis. Canad. M. A. J., 1960, vol. 82
[11] Sigurd Nitter-Hauge, Ivar Enge, 1973. Relation between blood lipid levels and angiographically evaluated obstructions in coronary arteries British Heart Journal. 35, 791-795.
[12]“Trick and Treat” (Barry Groves, 2008, p.63)
[13] Kannel WB, Gordon T., 1970.The Framingham Diet Study: diet and the regulations of serum cholesterol (Sect 24). Washington DC, Dept of Health, Education and Welfare.
[14] Ian A. Prior, M.D., F.R.C.P., F.R.A.C.P., Flora Davidson, B.H.Sc., Clare E. Salmond, M.Sc., and Z. Czochanska, DIP.AG., 1981. Cholesterol, coconuts, and diet on Polynesian atolls: a natural experiment: the Pukapuka and Tokelau Island studies. The American Journal of Clinical Nutrition 34, pp. 1552-1561.
[15] “The Great Cholesterol Con” (Dr Malcolm Kendrick, 2007, p.164-165)
[16] “Trick and Treat (Barry Groves, 2008, p.52)
[17] Scandinavian Simvastatin Survival Study Group, 1994. Randomised trial of cholesterol lowering in 4444 patients with CHD: the Scandinavian Simvastatin Survival Study (4S). Lancet; 344: 1383-1389
[18] http://www.westonaprice.org/cardiovascular-disease/dangers-of-statin-drugs
[19] Mukesh K. Jain, Paul M. Ridker, 2005. Anti-Inflammatory Effects of Statins: Clinical Evidence and Basic Mechanisms, Nature Reviews Drug Discovery 4. 977-987