
In my book the Clot Thickens, and in other blogs and lectures, I have stated that LDL cannot get through the endothelium (lining of blood vessel walls) and into the artery wall behind. So, the idea of LDL leaking from the bloodstream and into the artery wall is nonsense.
Recently, however, people have been bombarding me with papers and AI generated essays, stating that there are clear mechanisms that allow this to happen. Indeed, it does happen – so they say. The primary pathway is transcytosis.
So, LDL can get into the arterial wall past the ‘barrier’ of endothelial cells, and the impenetrable tight junctions between them? Am I wrong about this?
What is transcytosis?
I probably need to begin by explaining what transcytosis is, in as few words as possible. Unfortunately, not that few. With some other background bits and pieces.
The first point I want to make is that all human cells ruthlessly control what is allowed into, and out of them. If they lose this control they will die, almost instantly. It is, pretty much the definition of cell death. This ‘substance control’ occurs all the way down to the atomic level – the smallest size possible.
By which I mean that even ions (charged atoms) such as sodium, chloride, potassium and calcium have to pass through gates, or channels, to be allowed entry or exit. This is a tightly controlled process, requiring cellular energy, and a large number of complex mechanisms, including messenger molecules. The gates/channels are embedded in the cell membrane.
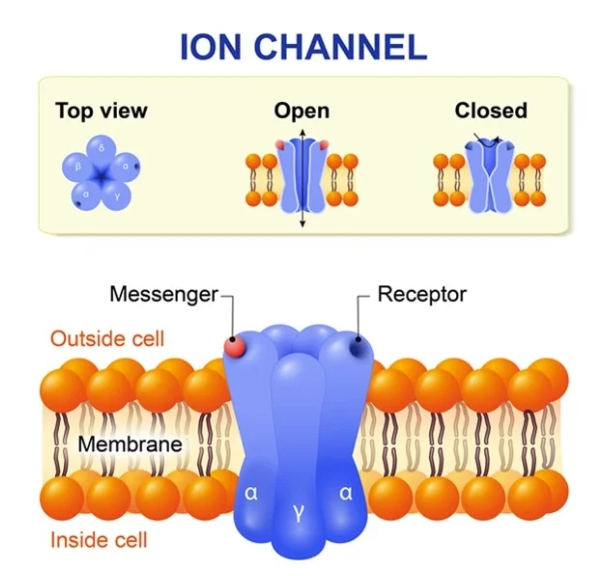
LDL molecules are thousands of times bigger than an atom. Ergo, there is simply no way they can force entry into a cell. [Terms and conditions apply, see under viruses].
Given this, how does LDL get from the bloodstream into endothelial cells – which it clearly does? The answer is that the cell manufactures a receptor, the LDL receptor which then travels to the cell membrane – where it embeds itself, ready to grab a passing LDL. This system was first identified by Goldstein and Brown – who won the Nobel Prize for their work in this area.
How does the receptor operate? Very simply, LDL has a protein attached to the side of it, the ApoB-100 protein, which is the ‘key.’ This key is perfectly designed to fit into the LDL receptor ‘lock’. Once locked on, the LDL molecule and the receptor are then pulled into the cell through a process known as endocytosis.
In essence the cell membrane wraps round the LDL/receptor complex forming a little lipoprotein sphere. Once inside the cell, the sphere is broken apart releasing its contents – including cholesterol and fat(s). And a few other things.
The next step is that the LDL receptor itself is – often but not always – broken down by an enzyme called, – wait for it – Proprotein convertase subtilisin/kexin type 9 (PCSK9). If the receptor is not disintegrated by this enzyme, it recycles back to the cell membrane, ready to lock onto another LDL molecule.
Most cells have thousands of LDL receptors stuck to the side at any one time. So, this is not a cottage industry, it is heavy-duty manufacturing.
That is how LDL can get into a cell. Through ‘endocytosis.’ ‘Endocytosis is the process by which cells absorb substances by engulfing them.’
[Endocytosis is also, mostly, how viruses get into cells. Viruses have proteins attached to their outer casing, such as the spike protein on Sars-Cov2. If this spike can find something to lock onto – in this case the A2 receptor – it will be ‘endocytosed’ and pulled into the cell. However, if there is no lock to be found, the virus cannot enter, and you cannot be infected by that virus. Interestingly viral particles and LDL molecules are pretty much the same size].
Below is the original diagram of the LDL receptor. It does not include PCSK9, because no-one knew it existed at the time.
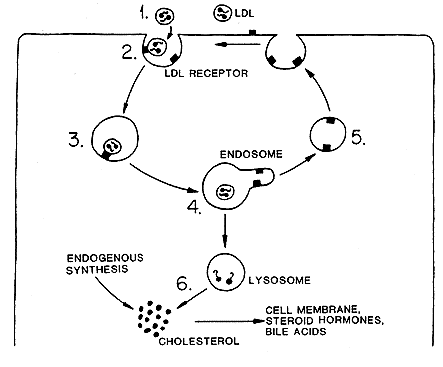
All of this is very widely accepted as fact – even by me.
So, there is no issue with getting LDL into a cell, which is the first step in transcytosis. Can LDL then be transported across the cell and popped out the back, into the arterial wall behind, a process called ‘exocytosis’? The full mechanism of transcytosis has three parts? Entry, then transport across the cell, then exit. Endo-trans–exo .. cytosis.
Problems
The first point I would like to make here is one of scale. If you were the size of an LDL molecule, the cell would be two kilometres across – approximately. Therefore, LDL is not going to simply drift from one side to the other, then bump into the cell membrane on the other side. Well, it might, but it would take a hell of a long time to do so.
Instead, it needs to be actively transported through the cell – in some way. Which means that, for transcytosis to work, the cell needs to have complex mechanisms within it designed to take LDL by the hand and lead it through the cell. Then exocytose it out the back. This is not some random – a million chimps writing Shakespeare – type of thing.
In super-simplified diagrammatic version, it would look something like this:
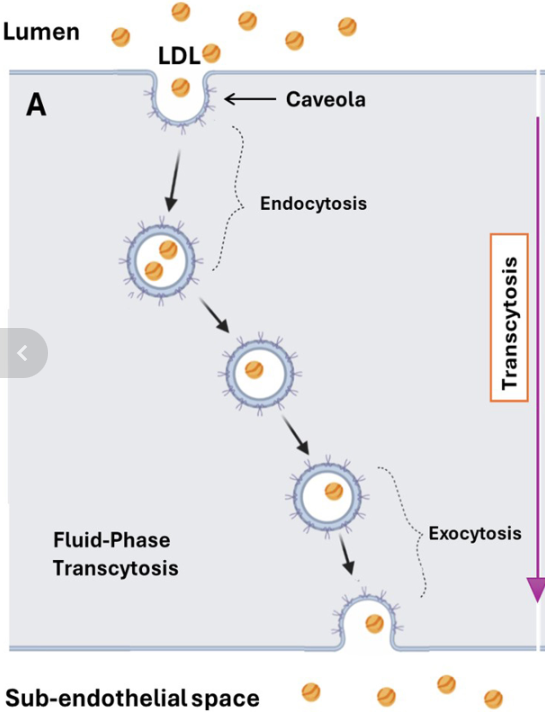
It is true that cells do possess mechanisms that enable them to transport various molecules from one side to the other. Then pop them out the back. Although, with LDL, the ‘how’ remains unclear, and vague. And the ‘why’ is even more uncertain.
As in, why would an endothelial cell go to all the trouble of absorbing an LDL molecule – then decide not to break it down – then transport it across itself before popping it into the sub-endothelial space behind? [A very narrow fluid filled space]
The only reason for this must be that the cells behind, in the deeper layers of the arterial wall, need cholesterol, and require it to be passed on to them. How would they communicate this need to the endothelial cell?
Well, for this to happen the cells in the arterial wall would need to synthesize a messenger molecule that travels through their cell membrane – then crosses the subendothelial space to lock onto a receptor on the outside of the endothelial cell membrane facing the arterial wall.
This would trigger another messenger molecule to say that the cells behind need LDL. So, could you please not break down all the LDL molecules you absorb and send them through to us instead. Yes, indeed, it’s a bit of a long and complicated message, but it could happen. The body acts in mysterious ways, its wonders to perform. Or something like that.
You may choose to believe that all such mechanisms, and messenger molecules, and the receptors for said molecules to lock onto exist, and have been fully identified … and have been seen operating in vivo. Or you may not.
But you can hopefully understand that we are not talking about some passive, ‘LDL moving down a concentration gradient from blood to arterial wall thing’ here. Sailing happily from the bloodstream directly into the arterial wall. You may also see why I am highly sceptical about all of this. Primarily, because it does not make any sense at all [see under vasa vasorum, discussed later].
Why so much recent focus on transcytosis?
Transcytosis has become an area of much research, and comment, recently. Why? Well, I like to think I may have been the cause of some of it. For I have stated in books, lectures, and writing that LDL cannot simply slip through – or past – the endothelial barrier. Thus, LDL cannot passively leak into the arterial wall, causing plaques. Ergo, the LDL hypothesis is wrong.
My position on this is, I believe, based on rock-solid logic. If LDL cannot navigate a way through undamaged endothelial cells, and the tight junctions that bind endothelial such cells together, then LDL cannot be the proximate cause of plaque development. Because it cannot get into the arterial wall to start creating a plaque in the first place.
I think this has, finally, been recognized as being a rather major obstacle to the LDL hypothesis. Whether through my work and writings, or for other reasons. Whichever, it now seems to have become more widely accepted that the concept of LDL travelling down a concentration gradient is scientific nonsense. And always has been. Sigh … head hits desk
Just to start with, a concentration gradient simply cannot shift things out of the bloodstream and into any cell. Because, sitting between the blood, and any cell, is a cell membrane. And, apart from anything else, you cannot have a concentration gradient between two ‘fluids’ if there is a solid barrier lying between them. No ‘gradient’ exists unless the barrier breaks down. Then things can travel about.
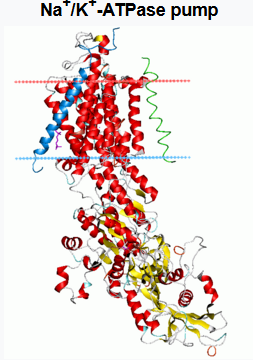
More relevant is the fact that cells are perfectly capable of shifting molecules up – against any ‘theoretical’ concentration gradient. They do this all day, every day. In fact, they do this millions of times a day. If this didn’t happen, we would cease to exist. Your heart would instantly stop beating, for example.
Here, from Wikipedia, is a short explanation of the sodium-potassium pump – which pumps sodium out of cells, and potassium in:
‘The sodium–potassium pump also known as Na+/K+-ATPase, Na+/K+ pump, or sodium–potassium ATPase) is a found in the cell membrane of all animal cells. It performs several functions in cell physiology.
The Na+/K+-ATPase enzyme is active (i.e. it uses energy from ATP) For every ATP molecule that the pump uses, three sodium ions are exported and two potassium ions are imported. Thus, there is a net export of a single positive charge per pump cycle. The net effect is an extracellular (outside the cell) concentration of sodium ions which is 5 times the intracellular (inside the cell) concentration, and an intracellular concentration of potassium ions which is 30 times the extracellular concentration.’
Yes, cells can pump potassium ions out and into the blood, or elsewhere, ‘against’ a massive concentration differential. And the potassium ions do not simply leak back in again, because they can’t. Because the cell membrane acts as a barrier to re-entry.
And just to boggle your mind a bit more there can be, up to, thirty million sodium-potassium pumps on an individual cell. [Nerve cells have the most, it’s how they pass electrical messages].
And an average endothelial cell will have around forty thousand LDL receptors on its surface. Which may give you some idea of how vital cholesterol is for healthy cellular function.
The point I am trying to emphasize here, perhaps over-emphasize, is that hundreds of different molecules move in and out of cells, under the control of complex cellular processes, requiring receptors and messenger molecules Nothing happens here that is not tightly controlled. And I mean nothing – viruses aside.
For many years there has been a horribly lazy assumption that, if the LDL concentration in the blood is high, it will simply move out of the bloodstream into the artery wall. Straight though the endothelial barrier.
Rather belatedly it has become more widely accepted that for the LDL hypothesis to work, there must complex mechanisms in place to allow LDL to travel though endothelial cells and into the ‘sub-endothelial’ space behind. And then get taken up into the arterial wall – in some mysterious way.
And lo, it seems, we now find that these mechanisms exist. Or do they? Small parts of the mechanism have been proven to exist, in some cells. As for the whole linked together system … nope. Not even close.
Some real-world stuff – PCSK9 Inhibitors and the vasa vasorum
PCSK9 Inhibitors
I hope to have made clear there are many (theoretical) areas where the transcytosis conjecture falls to bits. What about in the real world? For now, I am only going to look at two issues. I may look at others a bit later.
The newest LDL lowering drugs to hit the market are PCSK9 inhibitors. The thinking behind them is that, if you can block PCSK9, then fewer LDL receptors will be broken down. Therefore, more receptors will travel back to the cell membrane, more LDL will be absorbed by cells, and the LDL level will fall – thus reducing the risk of cardiovascular disease.
These wonder drugs include:
- Evolucamab (Repatha)
- Alirocumab (Praluent)
- Inclisiran (Leqvio)
It is true that they can reduce LDL in the bloodstream by, well over, fifty per cent. A much greater reduction than can be achieved with statins. But you may have just spotted a problem with the logic.
If more LDL is taken up by endothelial cells, then (if the transcytosis argument is correct) more ‘non-broken-down LDL’ will be available to be transported into the arterial wall behind – to form plaques. Ergo, you will be increasing the risk of cardiovascular disease, not reducing it.
So, do they work, or not? Well, I have looked at the studies and whilst they do not significantly increase the risk of cardiovascular disease, they have spectacularly failed to show much benefit. Here from a review of the FOURIER trial.
‘After readjudication, deaths of cardiac origin were numerically higher in the evolocumab group than in the placebo group in the FOURIER trial, suggesting possible cardiac harm.’ 1
Yes, reducing PCKS9 levels is amazingly effective at lowering of LDL, whilst slightly increased risk of cardiovascular death. Transcytosis argument proven, or disproved? I think disproved.
Perhaps more important, if not directly related to this discussion, is that they just managed to spectacularly disprove the LDL hypothesis. Sixty per cent lower LDL leading to an increased rate of cardiovascular deaths. Moving on.
The vasa vasorum
All arteries, of the size where plaques develop, have their own blood vessels lying within called vasa vasorum (blood vessels of the blood vessels). They supply nutrients to the arterial wall. It is their role in life.
Or, to look at this in another way, the cells in the arterial wall do not require access to the nutrients in the bloodstream that flow through the artery. If they need cholesterol from LDL molecules, for example, they can get it. Because it is travelling all around them in very small blood vessels which are purely designed to provide all the things the cells within arteries need.
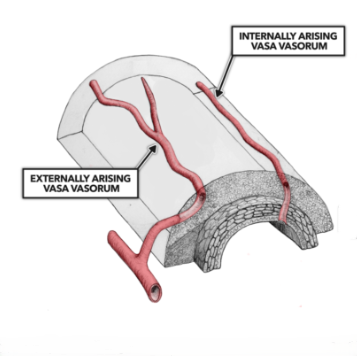
Yes, these vasa vasorum are lined with endothelial cells – as are all blood vessels. However, at the smallest ‘capillary’ size blood vessels the endothelium does not act as a barrier. They are, usually, leaky, and made deliberately so. Because, once blood arrives at its final destination substances have to be able move in and out of the blood freely.
Blood arriving at the kidneys, for example, has to unload waste products into each nephron – for ejection in the urine. Or the kidneys won’t work. And blood vessels in the liver are equally leaky.
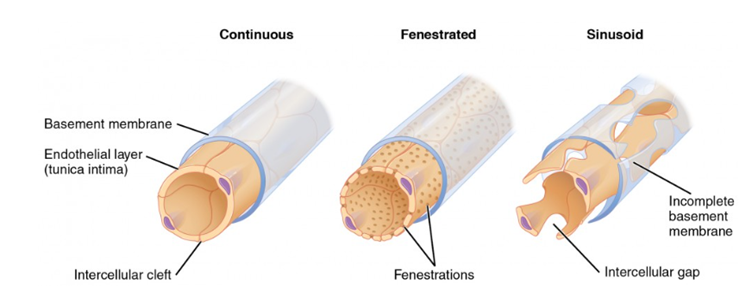
In fact, just to complicate things further, there are three different versions of capillaries [smallest blood vessels in the body]..
Continuous, where the endothelial cells are tightly bound together – and wrap back onto themselves. In addition, the basement membrane supporting them is also continuous. Continuous capillaries retain control of the passage of all molecules. They are found, mainly, in the brain, and this structure creates the blood brain barrier.
Then there are fenestrated capillaries, with small holes in the endothelium.
Finally, there are sinusoidal capillaries. These are very leaky, and are found within the vasa vasorum.
At this point, to my mind, the entire transcytosis argument becomes pretty much irrelevant. Why? Because LDL can get into the artery wall any time it likes, via the vasa vasorum. There is no barrier here. Entry by the back door, if you like.
So why have we become tangled up with complex arguments on transcytosis? If f there is a simple way that LDL can enter the artery wall any time it likes?
Because gentle reader, veins have more vasa vasorum than arteries. So, LDL can enter vein walls with even greater ease than artery walls. In addition the blood vessels in the lungs (pulmonary vessels) also have vasa vasorum.
And yet …
Plaques do not (except in exceptional circumstances) develop in veins or pulmonary blood vessels. I would say never, ever, but it can happen with, for example, Eisenmenger’s syndrome. [Very high blood pressure in the lungs].
All of this leaves the LDL hypothesis with a logical chasm to bridge. Starting with the first issue:
Why would LDL transcytosing into artery walls cause atherosclerotic plaques to develop, when LDL transcytosing into vein walls, and pulmonary arteries, does not?
Just in case you are wondering, the LDL level in veins is significantly higher than in arteries:
‘The present study’s main finding is that all lipoproteins are found in lower amounts in aortic blood when compared with peripheral venous blood’ 2
The ultimate ad-hoc hypothesis
I may seem to have come an awful long way to simply wrap back round on myself, and dismiss the entire concept. But transcytosis is presented in such a ‘clever’ way that it sounds entirely plausible. It is also made to sound difficult, and virtually incomprehensible. How can you argue against something that you do not understand? The fear of sounding stupid looms large.
The reality is that transcytosis sprang to life as an ad-hoc hypothesis. Here I quote AI on the matter of such hypotheses:
An ad hoc hypothesis is a provisional explanation or supplementary assumption added to a theory specifically to save it from being disproven or falsified by new, conflicting evidence.
Key Characteristics
- Crisis-Driven: It is invented to explain an anomaly that the original theory failed to anticipate.
- Lacks Independent Testability: It usually cannot be tested or verified on its own; its only evidence is the problem it attempts to excuse.
- No Predictive Value: It does not lead to new discoveries or broader scientific understanding
Transcytosis may seem, superficially, to provide an answer to the question ‘how can LDL move from the bloodstream into the artery wall – though an impenetrable barrier, and thus cause plaques.’ It happens by transcytosis. Phew, the LDL hypothesis is still correct.
But:
- LDL can get into the artery wall via vasa vasorum
- Thus, it can also get into vein walls and pulmonary vessel walls in the same way
- So, why does LDL not cause plaques in these blood vessels?
Which leads us back, as always, to the same, critical question. What is different about arteries in the systemic circulation* that makes them prone to develop plaques, when the same level of LDL has no such effect on, or in, any other vessels?
*The systemic circulation means blood leaving the left ventricle, before travelling round the body and arriving back at the right atrium. From where it is pumped into the right ventricle and then goes into the lungs, before arriving back at the right atrium – the pulmonary circulation.
As I hope you can see, invoking transcytosis to explain anything gets you nowhere, as all the blood vessels, large enough to develop plaques, have vasa vasorum in them that allow free passage of LDL.
Next ad-hoc hypothesis will be, I guess: “Why transcytosis only happens in arteries – at the exact point where plaques develop”.
Anyway, I do hope you have learnt something new. You may wish to explore what I have written in more detail. You may want to attack my reasoning. I welcome that. One of the main problems in science nowadays is that no-one debates anything.

In someone with secondary heart disease, someone who has had a stent or bypass, do you still no see benefit for these people in reducing LDL to slow the formation of new plaque?
Other question: what are your thoughts on the possibilities of restoring (in those who are dysfunctional) the conditions for efferocytosis so as to lower plaque and heart risk in people with cardiovascular disease?
Thank you, Dr. Kendrick, for continuing to bring logic, lucid reasoning, and hard facts to topics so often bereft of all three. Do keep up your terrific work
Quite a ‘technical’ Article…..one for the advanced in knowledge of the body’s arterial and venous systems.
Gor blimey! Whenever I read an intricate account of cell biology I simply thank God I was good at physics and maths and so didn’t have any temptation to enter the biological professions.
Of course it’s over my head; but always, when reading anything you write, I can FEEL your frustration and grumpiness – neither of which prevents humour. That’s a trio I recognize. 🙂
bravo ________________________________
I have read “The Clot Thickens” several times; I like to review it just before my annual visit to my cardiologist for defense purposes! I think this is an excellent article that summarizes a lot of the information in the book and ties it all together into a coherent, stand-alone, explanation of what can or cannot or doesn’t happen in arteries.
Thank you!
Firstly, Malcolm, the reason that there is no debate in science now is because all the little sheep follow each other and are scared to step out of line…..even just a little bit! Laziness comes into it too….tradition and training rule, and the vast majority can’t be bothered to use any more brain power once they have graduated!
So…….thank you for another fantastic piece of work. It must take you so long to read these hypotheses, analyse then write up your thoughts, with such clear diagrams too for us lesser mortals. Your books have travelled with me for years and have their own spot on my bookcase here in Sydney now, and I still dip into them from time to time.
Please keep swimming against the tide……we need you.
I would like to see how much relevance there is to the idea of heavy metals, nano-plastics and chemicals in the process of plaque formation. Also the role of seed oils and blood sugar in causing any of this. It seems that this particular malady is mostly a modern 20th and 21st century problem.
Not sure which book(s) but Kendrick names as a cause of endothelium damage and therefore a potential factor in atherosclerosis: some heavy metals (lead, primarily when it was used in petrol/gas), inhaled residue, as well as from smoking or other smoke (wood stoves, forest fires). It’s probably not a major risk, but don’t accidentally snort plutonium dust like that poor SOB does in Tom Clancy’s The Sum of All Fears.
I don’t recall him naming other metals, but certainly those, as well as the others you name, could be factors. I can’t recall him saying so, but certainly other heart books finger high glucose* and insulin spikes as very irritating to the endothelium.
*While Kendrick seems skeptical of these, other authors (Bowden & Sinatra) believe that oxidized or glycated LDL or other blood components to be suspects in the causes of atherosclerosis.
Very good, Doc. Being the ‘orrible skeptic that I am you have once again exposed how just-so stories get suitably aggregated to provide an all-round income. Medical wonders are just so, so rewarding, are they not? Of course the MSM and the nellies-on-the-telly just love a good drug deal. Please keep sniping!
Veins and arteries are not the same thing, so how can we be sure that LDL through vasa vasorum is not a problem?
Because veins develop plaques when used as bypass grafts in the heart
Veins have lower blood pressure than arteries. I assume veins will have the same pressure as other arteries when used as such, won’t they?. So I don’t find your answer fully convincing. What else would support your statement that LDL in vasa vasorum is not problematic?
Because when they transplant a vein they ensure they do not damage the vasa vasorum, as intact VV results in a better result
Thank you for that, for the piece and also for dedicating so much of your time researching and writing it. I must re-read “The Clot Thickens” again, now that I’ve been put back on a statin after a stent procedure (and considering dumping it.)
Hi, Dr Malcolm, Â Good stuff – keep it coming. Â I had never heard of PSK9 before this morning – then two come along at once – what do you make of this article: Â https://academic.oup.com/eurjpc/advance-article/doi/10.1093/eurjpc/zwag273/8690435?utm_medium=Email&utm_source=ESC+Global&utm_campaign=esc-newsletter-week23-2026&login=false https://academic.oup.com/eurjpc/advance-article/doi/10.1093/eurjpc/zwag273/8690435?utm_medium=Email&utm_source=ESC+Global&utm_campaign=esc-newsletter-week23-2026&login=false Â
  Kind regards,  George Hewitt CEng MICE   Â
I did a quick scan of the paper you linked. It argues that the drug(s) do indeed lower LDL and by a goodly amount. I noted (unless I missed it) the complete absence of any claims at all that health outcomes were improved. That is typical of the many similar papers I’ve looked at. We are to take it on faith that lowering LDL improves outcomes, forever and ever, amen. This is either a case of “the elephant in the room” or perhaps, “the elephant that should be there but is absent.”
Dr. Kendrick
excellent summary, have you considered the back door way that Subbotin proposed; vasa vasorum protruding into vessel muscles from the top, introducing all molecules available in the blood? He also pointed out the multilayered vessel epithelium, growing like a treerings for the first 25 years of life (under high pressure of an artery). Just thinking that inside multilayers have to be fed as well…
rgds JR
Still ploughing a lonely furrow, like john yudk
Just to say thankyou for your knowledge and time don’t ever give up!
Critical thinking! Scientific! Simply brilliant! Here’s the difference between science and “expert opinion”!
Is apoB concentration cumulatively during lifetime related to the risk of plaque formation and plaque burden?If so,even if it not the cause/the cause is endothelial injury and dysfunction,exposing underlying structures/,it might be a contributing factor
I would provisionally answer your question “Yes” but I don’t think ApoB number by itself is highly useful, any more than TC is. There’s a lot more nuance involved.
I think Kendrick is sceptical of ApoB by itself, yet other authors give it more credit. I’ll speculate a bit here (no expert) so take any of the following with a grain of salt. This is all based upon my kindergarten level understanding of the issues! Disclaimer done.
When you say “ApoB” what you really mean is “Apolipoprootein B-100” which is a marker unique to VLDL and its derivatives IDL (not mentioned in polite company, per the good Doctor) and more relevantly, LDL and perhaps Lp(a).
I’m assuming your quest would be the value of measuring, whether once or over a period of years, the average amount of ApoB in one’s body. What ApoB number would be measuring, and indiscriminately as far as I can tell, are the following:
The amount of VLDL (“TG”). It is well established that high VLDL is associated with CVD, most likely because it’s a marker for high carbohydrate consumption, which generally leads to metabolic syndrome and a host of allied conditions, including CVD.
LDL: in and of itself (perhaps) not harmful even when above official targets. The official church says “ High LDL bad.” Many researchers, but maybe not Kendrick, note that LDL comes in several flavors and that some of these may be harmful. I’ll leave out the case of Lp(a) since it has usefulness based on evolution; it may be a factor in CVD but I leave that for other discussion. Instead, let’s focus on the damaged LDL, which ostensibly can happen in many different ways. One is the particle size: large fluffy LDL is associated with a low-carb diet. They are clamed to not be harmful. Small dense LDL, on the other hand, associate with high-carb diets and are atherogenic. Finally, add in damaged LDL, either oxidized or glycated.
If the above reasoning is correct, then the ApoB number may be an indicator, but like LDL measurement alone, does not give enough information to make an accurate judgment of the atherogenic potential. From the ApoB number alone, you have no way of knowing whether VLDL, LDL or both are high.
I give you 9 out of 10 for that explanation
Fascinating article. Thanks
That’s all very well. So how come I had a massive heart attack th
I agree, and have read your Clot Thickens book. But you need to study the work of Gilbert Ling and Harold Hillman. All this crap about what cells do and what components comprise them is folly. This stuff is all hypothetical based on accepting that artifacts from the electron microscopy process are actually living working components of the cell. Just about all of medical science has been built on a foundation of shoddy science and outright fraud. People like Pasteur and Koch were certainly challenged in their day, but the monied interest (read Rockefeller and others) wanted to build a pharmaceutical behemoth, and they needed the evil, invading germ so they could kill it. That’s why their unproven ideas were codified. The history of all science in general is that the ideas that will make people money rule the day.
Absolutely brilliant, Malcolm! One of your best.
I have spent several hours debating AI on this very issue. It’s great fun watching Grok tie itself in knots trying to explain the inexplicable with an increasingly colourful word-salad. I highly recommend it as an activity for a wet afternoon.
As you say, the establishment solution appears to be to come up with arguments of increasing complexity in the hope that, eventually, we’ll just give up for fear of admitting ignorance.
keep up the good work.
Thanks to the good Dr. for writing this blog. I was one of apparently a host of readers who sent in questions about transcyosis and other sleight-of-hand that LDL reputedly does. I can’t take credit for it, but thank you Dr., as my questions were only several days ago!
A Quick Aside
I’m still fairly new to using the AI as an improvement over traditional Gogole search. If it matters, I’ve used the free versions of ChatGPT and Google Gemini. I’ve had fun asking innocent questions about (say) the process of how LDL causes atherosclerosis and let the AI take it from there. I then give it talking points I’ve learnt from Kendrick’s books or similar stores of forbidden lore. The AI is so obsequious! E.g. “You’re absolutely right! This is one of the most hotly debated issues in cardiology.” In fairness, the AIs did teach me some new terms and seem to provide references. Whether these have any merit, I cannot yet say.
A quick scan of the blog shows that he doesn’t cover these specified topics, so I’ll share (in brief!) what the AI claims are two major ways that LDL gets smuggled beyond the endothelium, it is alleged.
[The following section is an edited session from Google Gemini.]
LDL particles cross the endothelial cell barrier via two primary pathways: transcellular transport (transcytosis) and paracellular transport.
1. Transcellular Transport (Transcytosis):
The vast majority of LDL enters the arterial wall by going through the endothelial cells rather than between them. This is an active, vesicle-mediated process that relies on specific receptors to engulf and move the large LDL particles.
Receptor-Mediated Transcytosis
Two primary receptors on the luminal (blood-facing) surface of endothelial cells are responsible for binding LDL:
SR-B1 (Scavenger Receptor Class B Type 1): Research highlights SR-B1 as a primary driver of LDL transcytosis. It binds LDL and initiates its macromolecular transport across the cell.
ALK1 (Activin Receptor-Like Kinase 1): ALK1 is another crucial receptor that binds LDL independently of the traditional LDL receptor (LDLR) and facilitates its pathway through the endothelium.
The LDL Receptor (LDLR): While LDLR is vital for clearing LDL from the bloodstream into liver cells, its role in endothelial transcytosis into the arterial wall is secondary compared to SR-B1 and ALK1. [Sure it is! All cells have LDL receptors, and only the endothelial cell’s not only takes what it requires for cell function, but also schleps extra all the way across the cell.]
2. Paracellular [LDL sneaks in via the tight junctions – yeah right.]
[End Gemini long quote]
Elsewhere, I learnt the term pinocytosis: the cell more or less indiscriminately “drinks” a sample of whatever’s adjacent. This is ostensibly not receptor mediated.
You may, at your leisure of course, investigate these terms via your own search or AI session.
At one point, I backed the AI into a corner by pointing out that atherosclerosis is rarely found except at areas of high shear stress (e.g. the carotids, the coronary arteries) and didn’t this rather invalidate the “LDL smuggling” theory? The AI proffered the explanation was that the endothelium at those junctures is very stressed and in that dire condition is wont to suck up any LDL molecule that slams into the endothelial cell. (OK not that exact wording.)
In fact, that’s an excellent example of the fun one can have debating with the AI if one has read Kendrick’s books and is feeling a bit cheeky.
PCSK9 Inhibitors
After they learned I don’t “do” statins, my cardiology practice has, on two different occasions, recommended respectively Repatha or Leqvio. The latter was “new to me” so, using skills learnt from Doctoring Data I found a study or two and quickly determined that they really didn’t do much beyond lower LDL. I’ve politely declined these PCSK9 inhibitors. I admit I haven’t looked at Praulent.
Most, but not all cells are near a capillary
I’ve learnt the exceptions the hard way. Last year, inaugurating my arthritis, I developed a torn meniscus in my knee. While it’s true that part of the meniscus has blood vessels, my damaged part doesn’t – making any healing most improbable. I don’t claim to know the details, but it’s my understanding that such cells are bathed in synovial fluid. The cells survive even though they don’t have close access to blood vessels.
Even if you’re not interested in CVD, CVD maybe interested in you.
I’ve recently had imaging that at first sounded scary: my LAD artery (a rather important one!) went from 0 to 40% blockage in under six years. Fortunately, this is apparently still at the “moderate” stage. As is my CaC (50-ish). You can bet that I will be having this monitored closely. This is a bit annoying, as I don’t have many of the traditional risk factors for CVD. Age, maybe.
‘Last year, inaugurating my arthritis, I developed a torn meniscus in my knee. While it’s true that part of the meniscus has blood vessels, my damaged part doesn’t – making any healing most improbable.’
Around 2002 I was told the same thing, that the very tip of my meniscus had gotten torn sometime in the past and would someday require surgery, because having no blood supply, it would never heal. It hurt so bad at times that I could barely walk. The doctor prescribed an anti-inflammatory medication. Investigating that medication, I discovered that it could cause stomach problems so I never filled the prescription. Instead, I discovered a local farm/supplement producer had an herbal anti-inflammatory mixture designed for athletes.
I started taking the herbal mixture daily. After about six weeks, I noticed my knee didn’t bother me anymore. Neither did my elbows, which had started hurting from carrying my toddler. On occasion, I would run out of the mixture and a few days later the pain would return, only to disappear again once I started taking the herbs again. This happened enough times to provide evidence of efficacy.
Fast forward several years and I stopped taking the herbs. To this day, my elbow and knees are just fine.
After fighting off yet another cardiologist trying to kill my father, here you are in my inbox being brilliant again.
‘The Great Cholesterol Con’ saved my father’s life, Dr K. He was blue-lit (non-responsive) in 2011 and doctors stood around for 5 days trying to figure out what was wrong. Some even hinted at him having a heart attack even though there was zero evidence. We were left with some vague mumblings about him having had a ‘vasovagal episode’ and that was that.
Three years later, on the death of my mother, my father moved in with me. In terrible shape, I got him off the statins. All was well till our GP, after swapping rugby stories, rather charmingly cajoled him into getting back on them. Five days later, my brother calls: ‘He’s blue-lit again, non-responsive.’
‘It’s the damned statins!’ I said and hung up. Dad made another miraculous recovery. My brother tried hard not to look impressed. My GP still hasn’t read any of your books even though I said ‘pretty please’ and tied the stack up with a nice bow. And the world continues to turn wonkily.
(I omitted the bit where the GP tried to hand me the prescription, at which point my hands flew up in the air while I shouted, ‘It’s the devil’s pill! It’s the devil pill!’ This is between the two of you. I give up.’)
Thanks to you, my dad is 83 now—made from girders!—and his cardiologist is trying to sneak in a ‘statin replacement’. They have no shame.
Please know that I’m cheering you on from afar. You’re a total rock star.
Caitlin M
If arteries and veins are essentially the same yet plaques usually only form in arteries, or veins grafted to repair arteries, then surely the cause must be due to the difference in the application between the two?
The plaques form in the application that transports high pressure, oxygenated blood, surely this should be the starting point for investigation? Is the pressure/oxygen content a likely contributed?
The entire LDL scam (or the excuse for heart attacks) was invented to market statins and other heart drugs as well as prop up the need for stents. The virus scam (as well as germ theory) was invented to market endless vaccines and drugs upon which most of the existence of the medical mafia rests. Seems like the body is well guarded and designed to repel evil invaders at critical points and that includes imaginary viruses.
You’re entirely correct, of course. I would give you a summation of Kendrick’s (and many other’s): the diet-heart hypothesis predates statins by at least three decades: in the 1950s it was “cholesterol” that was bad, then saturated fat. Then LDL, probably. In the past several years it’s been the sub-types of LDL. Statins weren’t even marketed beforoe 1987. “Fat is bad” has been official (US) government dogma since mid 1970s. Not sure about UK.
In the UK, fat was ‘still good’ in the late 1970s especially for diabetics.
Three elderly friends of my parents, all sisters, were recommended to eat low carb/high fat by the GP after two of them became diabetic in their late 50s. Their oldest was in her early 60s and was considered borderline diabetic I think but was never diagnosed with the condition. She was wquite thin.
The overweight youngest and middle sister had normal lifespans and died in their mid 80s. The oldest sister lived to 89, having never been diagnosed ‘diabetic’.
I really enjoyed that. It brought to mind learning about aquaporins many years ago that evolved with a neat way of blocking hydrogen ions. Indeed, cells are very strict about what can get in and out. I forget where I came to know this, maybe even your book, but autopsies of young people showed that cholesterol moved from the outside inwards, towards the intima.
After posting something many other thoughts pop up. My usual argument against the official cholesterol/CVD hypothesis is that cholesterol has been part of animal cells since animal cells and there have been hearts and blood vessels for about half a billion years, plenty of time for natural selection to make the two work together. On the other hand, if you are one who believes in intelligent design, you have a mighty big problem.
There are no doubt many arguments against intelligent design, but my favorite is the “recurrent laryngeal nerve (RLN) is a major branch of the vagus nerve (Cranial Nerve X) that controls the muscles of the larynx (voice box)” (Source: Google). Consider the extreme case of the giraffe. Design, maybe. But intelligent? Well, sure doesn’t seem so, unless running a nerve from the brain, down to the heart, and back to the larynx is good design. Even the German’s aren’t that elaborate (“Why use five parts when fifty will do?”)
“…if you are one who believes in intelligent design, you have a mighty big problem”.
Ain’t that the truth. Since the Theory of Evolution was published and some of the details filled in, it has met the requirements of Occam’s Razor. It is the simplest explanation of everything.
As for God and the spiritual world, Laplace’s reply to Napoleon seems apt. “Sir, I didn’t require that hypothesis”.
How does it work with Lp(a) then?
Sent from Outlook for iOShttps://aka.ms/o0ukef ________________________________
That would take several pages to explain. Or, you could read the clot thickens, where it is explained
I’ve read it half a dozen times and refer to it weekly, I’ve measured my Lp(a) and it’s 96.4, which is ok but not brilliant. Beyond taking 3g of vitamin C per day I don’t know what to do! Don’t stress it I suppose (apoB high too)
Sent from Outlook for iOShttps://aka.ms/o0ukef ________________________________
Look up the Pauling protocol. Vitamin C (ascorbic acid form) should be 5-10 grams/day in divided doses. Add 5 grams of L-Lysine and 1-2 grams of L-proline. These three make Lp(a) less likely to cause problems.
I asked Gemini about your arguments, its response:
In chemistry and biology, the Law of Mass Action states that the velocity of a chemical reaction or a biological process is directly proportional to the concentration of the reacting substances.
When applied to cardiology, it means that the rate at which plaque forms is directly driven by the total number of plaque-causing particles bouncing against your artery walls.
Dr. Kendrick argues that LDL cannot enter the arterial wall unless an injury or a blood clot damages the endothelial cells first. The law of mass action exposes the flaw in this “injury-only” thesis:
This is simply unevidenced unscientific gibberish. Unworthy of any reply
’Even in perfect soil, if you flood it with an overwhelming number of seeds, things will grow’ ????
Presumably this is where they get the word-salad from.
Amazing. If you plant seeds in perfect soil, things will grow. Who’d ever have guessed such a thing might happen. A complete surprise to all gardeners.
Many thanks Malcolm.
The arguments they put forward distract from obvious things that anyone could understand. E.g. cholesterol needs to be in damaged blood vessels because it repairs. The general public is never told this.
And sending this in response to Carol’s posting (June 3):
COMPLIANCE
Like sheep that know not where they go
Nor care why so, since
There is safety in numbers
We think alike, if thinking be
A just word for apathy
That leads to strife.
Tish
From Dr James Lefanu’s book ‘Too many pills’.
“My first intimation of the scale of this debacle came just over a decade ago in a letter from a reader of my Telegraph column who, eighteen months earlier, had required a major operation to repair an aortic aneurysm – ballooning of the main blood vessel in his abdomen. He recovered well but had subsequently gone progressively downhill, becoming increasingly decrepit, immiserated by muscular aches and pains that his doctors were unable to explain. He was determined at least to make it to his son’s wedding in Hawaii – not an easy journey, requiring a wheelchair at the several transfer stopovers. Reaching his destination, he realised he had forgotten to pack the cholesterol-lowering statins prescribed after his operation but was so markedly improved by three weeks not taking them, he was able on his return to walk unaided back through Heathrow.
This account of his near miraculous recovery was promptly corroborated by hundreds of others describing how their impaired mobility, lethargy, poor concentration or insomnia often misattributed to anno domini (‘what can you expect at your age?’) had similarly resolved after discontinuing for one reason or another their medicines. And as time has passed, so the toll of polypharmacy has become ever more apparent. For those who are otherwise fit and healthy, the onset of side effects soon after initiating treatment will indicate the cause. The difficulty arises when these symptoms are more insidious or their doctors obtusely refuse to acknowledge the drugs might be responsible – as with the following from a woman whose 71-year old husband is taking eight pills in the morning, five in the evening. “He has lost all confidence”, she writes, “He gets up in the morning with no energy unable to work in the house or garden. When our grandchildren visit, it is all too much for him. His life stretches out with no hope of improvement. On enquiring about the necessity for taking so many medicines, the doctor said he would not be here if he stopped taking any of them.”
From a synopsis of Dr James Lefanu’s book ‘Too Many Pills’.
“My first intimation of the scale of this debacle came just over a decade ago in a letter from a reader of my Telegraph column who, eighteen months earlier, had required a major operation to repair an aortic aneurysm – ballooning of the main blood vessel in his abdomen. He recovered well but had subsequently gone progressively downhill, becoming increasingly decrepit, immiserated by muscular aches and pains that his doctors were unable to explain. He was determined at least to make it to his son’s wedding in Hawaii – not an easy journey, requiring a wheelchair at the several transfer stopovers. Reaching his destination, he realised he had forgotten to pack the cholesterol-lowering statins prescribed after his operation but was so markedly improved by three weeks not taking them, he was able on his return to walk unaided back through Heathrow.
This account of his near miraculous recovery was promptly corroborated by hundreds of others describing how their impaired mobility, lethargy, poor concentration or insomnia often misattributed to anno domini (‘what can you expect at your age?’) had similarly resolved after discontinuing for one reason or another their medicines. And as time has passed, so the toll of polypharmacy has become ever more apparent. For those who are otherwise fit and healthy, the onset of side effects soon after initiating treatment will indicate the cause. The difficulty arises when these symptoms are more insidious or their doctors obtusely refuse to acknowledge the drugs might be responsible – as with the following from a woman whose 71-year old husband is taking eight pills in the morning, five in the evening. “He has lost all confidence”, she writes, “He gets up in the morning with no energy unable to work in the house or garden. When our grandchildren visit, it is all too much for him. His life stretches out with no hope of improvement. On enquiring about the necessity for taking so many medicines, the doctor said he would not be here if he stopped taking any of them.”
The main engine driving polypharmacy is, if predictably, Big Pharma which since the 1980s has deployed its immense wealth and influence to blur the boundaries between the normal and abnormal, extrapolating in devious ways the undoubted merits of their drugs for the relatively few with, for example, markedly raised blood pressure or cholesterol levels to the vastly greater number in whom these indices are only marginally raised, if at all. Who would know, given the current enthusiasm for mass medication, that the benefits of commonly prescribed drugs are systematically exaggerated fiftyfold or that in certain instances 98% of those taking them gain no benefit from doing so?”
Yes, I know James well. Good man, fighting the incoming tide by my side. Unfortunately, money rules this world. And my medical colleagues are, not quite sure what the word is… Incurious?
Hi Malcolm, Thank you for keeping me in the loop with your emails First of all a few big Congratulations are in order: I want to congratulate you on your recent High Court success – delighted for you and your family – waiting almost six years for a verdict must have been extremely stressful. Also Congratulations on your recent Retirement – welcome to the club ! and I wish you many years of fruitful retirement. Thank you for writing your books on statins and health issues ( I have three of them ! ) Finally, I paid to view a webinar ‘ We love our Heart ‘ run by a guy called Mark Felstead where you were named to speak on May 16 last. Would you please confirm if you were asked to be one of the speakers on this webinar as I have a feeling now that it was all a scam ( hope I am wrong ) and your good name was exploited to entice people to buy this product. Dr Asheem Mulhotra and Ivor Cummins were also mentioned in this ‘ We love our Heart ‘ webinar. This webinar was supposed to run over two days May 16 – 17 and as I say you were supposed to be one of the speakers on the 16th. I tried all the links this Mark Felstead ( or whatever his name is ) gave me and none of them worked !! Then he said he would forward the recordings within minutes !! – still waiting ! If you didn’t know about this ‘ We love our Heart ‘ webinar then It confirms for me that it was all a scam and I feel you are entitled to know your name was used in the advertising of same. My apologies for intruding in your retirement space. Again every good wish for the future. Yours Sincerely Ted Coughlan
The webinar got moved about, but did happen and I did talk and answer questions. Mark Felstead is a good man who I like, who I think is truly trying to help people, and also pays the contributers to his conferences – not much, but a bit. My luxury yacht is still pending.
As for Aserm and Ivor. I know them both well, and we are friends. I do necessarily agree with everything they say about everything, but they are in my short book of good guys.
Here’s my dummy-spit that answers all questions.
The purpose of lipoproteins is to transport insoluble nutrients and biochemicals from the liver to cells as and when needed; on the return journey, insoluble waste is returned to the liver for elimination.
In writing my book, The Fact-Free Science of Cholesterol, I point out that LDL is a large particle consisting of an apolipoprotein with large spongy free cholesterol and phospholipid molecules covering the outside plus a belly-full of other insoluble nutrients and biochemicals such as cholesteryl esters, triglycerides, insoluble vitamins, CoQ10 and more. HDL particles are exactly the same as LDL particles except that they have less cargo.
Questions:
Why is it that large but nasty LDL particles breach the lining of arteries but smaller not-nasty HDL particles do not?
Why is it that when nasty LDL particles breach the artery epithelium, it drops only its cholesterol load?
Why not the phospholipid load?
What happens to the LDL particle and the remainder of its cargo?
Answers
The simple answer is: it doesn’t happen.
More Questions:
Why would the body that is smart enough to keep us in peak health if we feed it all the nutrients it needs, then start making extra cholesterol that it doesn’t need, risking atherosclerosis?
Then of course, it has to create HDL as a means of mopping up this excess cholesterol. Does that sound like the result of thousands of generations of evolution to you?
Has the world gone mad that it has swallowed this rubbish science as real?
Looking at the cholesterol issue from the lipoprotein point of view, that is, transporting insoluble stuff through the bloodstream, together with the basics of physiology and biochemistry, it soon becomes clear that the whole thing is a fabrication:
Fact: The so-called lipoprotein density sub-fractions, VLDL, LDL and HDL are not real, they are artefacts of laboratory measurement (bunging just about anything into a centrifuge will separate it into layers of different density).
Fact: The cholesterol on lipoproteins is the same regardless of lipoprotein density.
Fact: There are no LDL receptors because there is no LDL per se. In fact, there are no lipoprotein receptors, there are only enzymes and receptors for the individual components being transported on lipoproteins.
I really love the title of your book Judy.
Fascinating and helpful to have these insights Dr Kendrick such valuable research. You write so clearly and accessibly I must say its a pleasure to read your pieces.
Very interesting and compelling. Based on the knowledge that you have gained what is likely to be causing the plaque buildups that create an issue for so many people, even those who can be extremely fit and relatively slim?
Beautifully written.
I first encountered transcytosis in social media. Which should have been my first warning. The term was promoted by physicians who condescended to inform the rest of us that (1) ApoB-containing lipids and especially LDL are causative, (2) all other possibilities are risk factors but not causative, (3) transcytosis is how LDL travels through the endothelium, (4) there is no limit to how low LDL should be driven with drugs, diet and lifestyle changes because (5) cells can make all the cholesterol they need endogenously.
I did a deeper dive into transcytosis, hoping to expand my primitive understanding of all things cardiovascular. Transcytosis was found in rabbits and rats in the early 1980s. 45 years later transcytosis has never been confirmed in vivo in human subjects. If they’re so certain that transcytosis is a thing in human endothelial cells, then why not eliminate all doubt?
The adult human body has between 28 and 37 trillion cells. Most cells have thousands of LDL receptors. Why? If cells can make cholesterol endogenously (which is a fact), then why does the adult body have LDL receptors numbering in the quadrillions? I’m pretty sure the answer is related to energy efficiency within the cell. But the more important question is, what happens in the long term when trillions of cells are forced to produce much of their own cholesterol instead of simply importing ApoB-containing lipids from the blood stream? Thanks to millions of human guinea pigs, we will have the answer before much longer.