
16th September 2021
Bridging the gap between cardiovascular disease and COVID19
[Where two diseases meet]
Having announced that I will not discuss COVID19 anymore, I am about to do so – at least in part. Yes, you may now be thinking… how can we believe anything this man says?
However, I do have an excuse for this. Because, as part of my transition back to more familiar waters, I am going to look at the links that COVID19 has to cardiovascular disease… my life-long obsession.
The reason is that I have found it amazing how two apparently unrelated diseases can be linked so closely, and greatly increase your knowledge of both.
I will start with a quote that I would like to you read slowly, and carefully, taking a little time to think about – if you can get through the jargon.
‘Host defense against infection is based on two crucial mechanisms: the inflammatory response and the activation of coagulation. Platelets are involved in both hemostasis (blood clotting) and immune response. These mechanisms work together in a complex and synchronous manner making the contribution of platelets of major importance in sepsis. This is a summary of the pathophysiology of sepsis-induced thrombocytopenia*, microvascular consequences, platelet-endothelial cells and platelet–pathogens interactions.’ 1
*thrombocytopenia = drastic fall in platelet levels (small cells that conduct the entire blood clotting orchestra).
Yes, as you may have noticed, this passage says nothing about COVID19. On the face of it, it has nothing to do with cardiovascular disease either. It also contains a lot of jargon which most people without a medical background will struggle to understand. To me, however, it is fascinating, as it opens an entirely new way of thinking about critical disease processes.
What these researchers are saying, in the typically impenetrable prose of medical writing, is that the immune system, and the blood clotting (coagulation) system, have been designed to work together to fight off infective agents. Indeed, from an evolutionary perspective, they started off as the same thing. As discussed in an article in the Journal ‘Immunity’. ‘The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1α by Thrombin.’
‘Ancient organisms have a combined coagulation and immune system, and although links between inflammation and hemostasis (blood clotting) exist in mammals, they are indirect and slower to act. Here we investigated direct links between mammalian immune and coagulation system….The identification of a direct link between the coagulation system and the activation of the IL-1α* inflammatory cascade raises important questions.’ 2
*Interleukin 1 alpha (IL-1α) also known as hematopoietin 1 is a cytokine** of the interleukin 1 family that in humans is encoded by the IL1A gene. In general, Interleukin 1 is responsible for the production of inflammation, as well as the promotion of fever and sepsis. [Which is why you get hot and shivery when you get infected]
**a cytokine is a small protein that normally passes messages from cells to other cells and the immune system. Cytokines are key players in the immune response to infections, and there are many of them.
Anyway, put at its simplest. If you become infected (with almost any micro-organism,) you are far more likely to produce blood clots. Why? Well, it is probably because serious and life-threatening infections will often enter the body through a wound, or damage of some sort. Therefore, it makes sense that the body tries to seal off such wounds, or entry points, with a blood clot. This will not only stop the bleeding, but it will also trap the invading bacteria and viruses to prevent them spreading.
At which point the immune system gets to work on the trapped micro-organisms. Indeed, what better way to neutralize a virus, or bacteria, than by wrapping it up inside platelet fibrin complexes – two of the main constituents of blood clots?
At this point you may well ask, so what has this to do with cardiovascular disease, atherosclerosis and atherosclerotic plaques? Well, as the same paper goes on to say:
‘Many diseases are driven by the interplay between coagulation and inflammation. Inflammation drives atherosclerosis and IL-1α can play a dominant role independent of inflammasomes suggesting another mechanism activates IL-1α. Plaques contain thrombin-antithrombin complexes and show fibrin localized throughout, implying thrombin activation occurs throughout atherogenesis. Thus, p18 IL-1α might drive atherogenesis.’ 3
In super-short version:
Infection → inflammation + coagulation → (if regularly repeated) atherosclerotic plaques = cardiovascular disease
I find it a remarkable coincidence that I was studying the impact of infectious agents on cardiovascular disease when the COVID19 tsunami broke upon the world. Then I started delving into what the Sars-Cov2 virus does to a wide range of physiological systems. It opened doors into new passageways of thinking, and research, that I never even knew existed.
Primarily, that there is a tight connection between the blood clotting system and the immune system. Who knew? Well, some people obviously did, because they were researching it and writing about it. However, until COVID19 came along I didn’t have the faintest idea. I hadn’t even thought to connect the two processes.
Yes, I already knew that infectious diseases, such as Influenza, could greatly increase the risk of a fatal blood clot in the days and weeks following infection. I knew that sepsis (bacterial infection of the blood) causes damage to endothelial cells that line all blood vessels, triggering small blood clots all around the body. A condition known as Disseminated Intravascular Coagulation (DIC), which is the primary cause of death in sepsis.
I also knew that ‘inflammation’ of the blood vessels, a condition often known as vasculitis, could greatly increase the risk of cardiovascular disease. Vasculitis essentially means damage of the endothelium (the layer of glycocalyx, and endothelial cells, that line all blood vessel walls).
The impact of vasculitis on cardiovascular disease is highlighted by the fact that the form of vasculitis associated with Systemic Lupus Erythematosus (SLE) a.k.a. ‘lupus’ can increase the risk of death from cardiovascular disease by – up to – 4,900% in young women. 4
Indeed, all the vasculitides – plural of vasculitis – can greatly increase the risk of CVD, and thrombosis (blood clotting):
‘The relationship between inflammation and thrombosis is not a recent concept, but it has been largely investigated only in recent years. Nowadays inflammation-induced thrombosis is considered to be a feature of systemic autoimmune diseases such as Systemic Lupus Erythematosus (SLE), Rheumatoid Arthritis (RA), or Sjögren Syndrome (SS). Moreover, both venous and arterial thrombosis represents a well-known manifestation of Behçet syndrome (BS).’ 5
Then, of course, along comes COVID19, which brought a number of these strands into tight focus. It became clear that COVID19 also links infection + coagulation + vasculitis.
How so? Well, it was rapidly established that COVID19 enters cells by linking onto a receptor known as the ACE2 receptor (Angiotensin Converting Enzyme 2 receptor), before being dragged into the cell.
ACE2 receptors form an important part of the enormously complex Renin Aldosterone Angiotensin System (RAAS). Sorry, this is yet another strand, but please bear with me for a while, because it is important.
What is the Renin aldosterone angiotensin system? Well, keeping it super-simple, the RAAS controls blood pressure. If your blood pressure drops the RAAS kicks into action. [It also kicks into action if sodium levels fall, but that is an entirely different world of discussion]. The RAAS forces the heart to pump harder, it constricts blood vessels, it drives the kidneys to keep a hold of sodium and water etc. etc.
Although there are all sorts of hormones involved in the RAAS, with feedback and amplification loops here and there, they basically all end up triggering the conversion of a hormone called angiotensin I to angiotensin II. Angiotensin II is the active hormone that locks onto receptors in various organs, causing them to do their blood pressure raising thing.
[If you block the conversion of angiotensin I to angiotensin II, you will lower the blood pressure. This is what the class of drugs known as ACE-inhibitors do. They inhibit the enzyme that turns angiotensin I into angiotensin II. Which means that they are called angiotensin converting enzyme inhibitors. This reduces the amount of angiotensin II in the blood, and stops the heart rate increase, the blood vessel contraction, and suchlike. These drugs are widely prescribed]
As you might imagine therefore, ACE2 receptors are present in high numbers on the surface of membranes of cells that play a role in the RAAS. Basically, any cells involved in blood pressure control.
A large number are found in the cells in the lungs, because the lungs are where Angiotensin I (the inactive pro-hormone) is converted to Angiotensin II – the active form. Why does this conversion occur in the lungs, not the kidneys or liver? No idea. Something to do with evolution probably.
ACE2 receptors are also found in the cells that line all blood vessels – the endothelial cells. Why? Because angiotensin II links to these receptors to create messages commanding blood vessels to constrict – thus raising the blood pressure.
[In fact, sorry to add yet another complication, ACE2 receptors represent part of the ‘control feedback system’ for RAAS. When activated, ACE2 receptors block the effects of angiotensin II. They are ‘anti-angiotensin II’ receptors, if you like. They work to keep the effects of angiotensin II from running out of control. However, they are still an integral part of the RAAS system, and a critical part of the negative feedback loop to control blood pressure. Thus, wherever you have an ACE-receptor, you will also have an ACE2 receptor. Yin and Yang].
Why is all of this important, you may ask. Because it explains which cells are going to be most damaged by COVID19, and why. Essentially, the cells that are most damaged will be the cells that play a role in the RAAS. They are damaged because they have ACE2 receptors on their membranes.
Without this receptor, it is impossible for a cell to be infected by Sars-Cov2, and no damage can occur.
Years ago, I was looking at the Ebola virus. I found out that this virus gains entry through a protein stuck to the cell membrane known as the CCR5 protein. As with COVID19 and the ACE2 receptor, Ebola must find something on the cell membrane to link onto, before it can gain entry to the cell. A lock and key if you like. If the lock doesn’t fit the key – there can be no entry for the virus.
It was found that some people have a variant of this protein known as the ‘CCR5 Delta 32 mutation’. Because this protein has a different structure to the normal CCR5 protein, the Ebola virus cannot link to it. Therefore, it cannot enter any cells. Which means that people with the CCR5 Delta mutation cannot become infected with Ebola. Or at least, it cannot enter any cells in the body, so it cannot multiply, so it cannot cause any damage.
It is of interest that HIV also enters cells using the CCR5 protein, and people with the CCR5 delta 32 mutation cannot be infected with HIV either.
Anyway, trying desperately to bring things back together… deep breath. Once inhaled, COVID19 gets into lung cells using the ACE2 receptor – creating lung damage. It gets into kidney cells – creating further damage. It gets into heart cells (myocytes, pericytes) – causing even more damage. It gets into endothelial cells – creating vasculitis. It also stimulates the coagulation system into action – as almost all infectious agents do.
If you survive the initial lung damage – which most people probably will do – then the thing you need to start worrying about is the vasculitis/blood clotting that will be triggered throughout the rest of the body. This will all be worsened by the fact that infected endothelial cells will be sending out cytokines (distress messages) to the immune system. Stating, simply. ‘I am infected, come and kill me and the virions within.’
This, then, is the basis of the ‘cytokine storm’ which you may have read about with COVID19. Ironically, the body’s own defence system, the immune system, can become the very thing that kills you with COVID19. It revs up, starts attacking the infected cells, and creates major problems such as myocarditis (inflammation/damage to heart muscle). Kidney damage/failure, and a more widespread severe vasculitis develops as the endothelial cells are machine gunned by their own side.
All of this creates widespread blood clotting, which was recognised quite early on. Here from the paper ‘Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications.’
‘Reports of widespread thromboses and disseminated intravascular coagulation (DIC) in patients with coronavirus disease 19 (COVID-19) have been rapidly increasing in number. Key features of this disorder include a lack of bleeding risk, only mildly low platelet counts, elevated plasma fibrinogen levels, and detection of both severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and complement components in regions of thrombotic microangiopathy (TMA). This disorder is not typical DIC. Rather, it might be more similar to complement-mediated TMA syndromes, which are well known to rheumatologists who care for patients with severe systemic lupus erythematosus or catastrophic antiphospholipid syndrome.’ 6
Again, much jargon. However, the final sentence which provided me with the intellectual equivalent of sipping a twelve-year-old malt whisky… Roll it around the palate with deep pleasure. Please read again, and think about it:
‘Rather, it might be more similar to complement-mediated TMA syndromes, which are well known to rheumatologists who care for patients with severe systemic lupus erythematosus or catastrophic antiphospholipid syndrome.’
On the face of it, a rather boring sentence. What it is telling us, however, is that with COVID19 we are looking at almost the same pathological process as seen in Systemic Lupus Erythematosus (SLE), with an added dash of antiphospholipid syndrome.
Lupus, as mentioned before, causes vasculitis, because the immune system attacks endothelial cells. It is made worse when the person also has antiphospholipid syndrome (sometimes called Hughes’s syndrome).
Phospholipids essentially, are cell membranes. Two layers of phospholipids stuck back-to-back like Velcro. Within this bi-layer of phospholipids are various channels and gates and receptors and (as you may have noticed), lots of cholesterol – which stabilises the cell membrane. No cholesterol, no cell membrane, it simply falls apart.
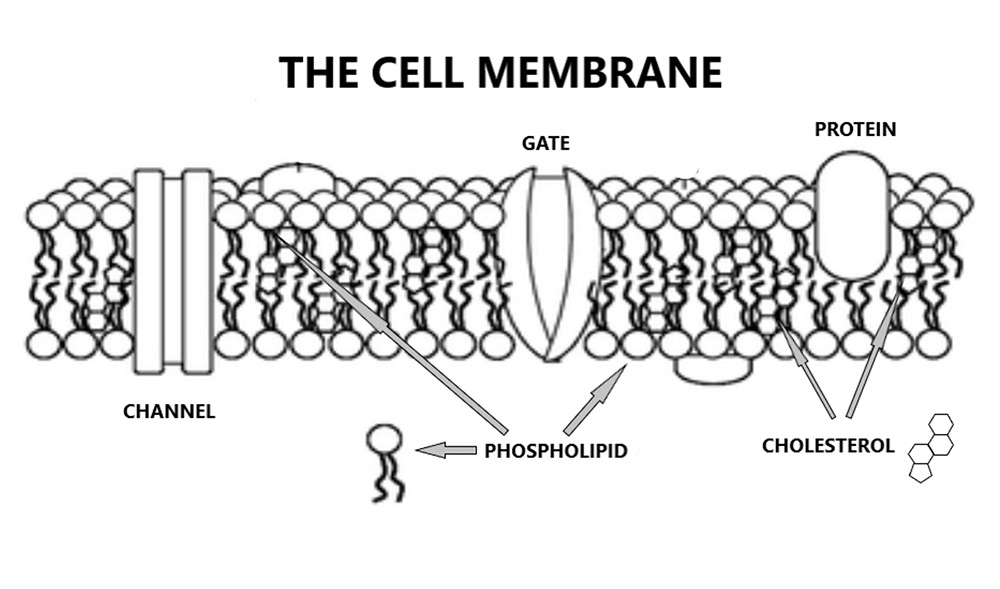
Getting back to anti-phospholipid syndrome, it means exactly what you would think it means. The immune system starts to attack the phospholipid bi-layer that makes up the endothelial cell membrane, it becomes an ‘anti-phospholipid system’. This creates damage, the damage exposes the underlying clotting factors, and you end up with blood clots forming on blood vessel walls. Thrombotic microangiopathy (TMA).
Thus SLE/antiphospholipid syndrome, and COVID19, although they are completely different diseases, can create almost the same damage. The immune system and clotting system combining – along with severe endothelial disruption. This is also, almost certainly, why some children develop a severe vasculitis following shortly after the acute phase of COVID19 infection.
Here, from the article ‘COVID-19-associated vasculitis and vasculopathy.’
‘COVID-19 is a SARS–CoV-2 syndrome that can involve all organs, including the circulatory system. Endothelial cell inflammation occurs within arteries, arterioles, capillaries, venules and veins and contributes to pathological events; including tissue hypoperfusion, injury, thrombosis and vascular dysfunction in the acute, subacute and possibly chronic stages of disease. Beyond re-writing the textbooks that hence will include SARS–CoV-2 as a causal pathogen for multi-bed vasculitis, the data will show that it is a new category of systemic vasculitis forever captured in the annals of medicine.’ https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7373848/
Look, I understand this is all complex, and I have taken you through it all at a bit of a rush, but I was hoping to give you a sense of my scientific excitement. When COVID19 hit, I was looking at vasculitis and how it caused cardiovascular disease. Here, are the very words I was writing.
‘Vasculitis means damage and inflammation to the blood vessels. Vascular = blood vessels; ‘itis’ = inflammation. As in tonsillitis = inflammation of the tonsils, or appendicitis = inflammation of the appendix.
There are many, many different sorts of vasculitis, and they all have impossible to remember names. However, I do love them, as they are so evocative of a bygone era in medicine. Here are several of them, not including systemic lupus erythematosus or rheumatoid arthritis:
- Polyarteritis nodosa
- Waldenström’s macroglobulinaemia
- Sjogren’s disease
- Giant cell arteritis
- Behcet’s disease
- Buerger’s disease
- Churg-Strauss syndrome
- Cryoglobulinemia
- Granulomatosis with polyangiitis
- Henoch-Schonlein purpura
- Kawasaki disease
- Takayasu’s arteritis
This is Harry Potter stuff. Wave your wand about and exclaim…’Vasculitis obliterans!’ Actually, that is another form of vasculitis. The reason why they don’t all appear on Qrisk3 is because many of them are considerably rarer than hen’s teeth. In addition, they not widely recognised to increase CVD risk – although they all do. If you choose to look.
Apart from increasing the risk of CVD, another characteristic they have in common is that they are also, what are termed as auto-immune conditions. ‘Autoimmune’ describes the situation whereby the body decides to attack itself….’
Immune system + vasculitis + coagulation.
How strange that a virus would come along and create an almost perfect model to highlight this world, I thought.
As a sign-off, I did wonder what it was with COVID19 that so directly stimulated the blood clotting system. As it turns out, it appears to be the spike protein itself. Here, from the paper ‘The unique characteristics of COVID-19 coagulopathy.’
‘Thrombosis is a major pathological driver in COVID-19. Evolving evidence suggests that in addition to the activated leukocytes and derangement of antithrombotic property of endothelial cells, hyperactive platelets participate in thrombogenesis. The direct and indirect effects of SARS-CoV-2 spike protein on platelets stimulate the release of platelet factor 4. The spike protein also upregulates inflammation and coagulation through the binding to ACE2 on macrophages/monocytes, lung epithelial cells, and possibly vascular endothelial cells, reactions that lead to micro and macro circulatory clotting known as CAC (COVID19 associated coagulopathy).’ 7
Yes, the spike protein. This, it appears, is the key antigen, the key driver of the immune/thrombotic system in COVID19. This is the factor that can lead to blood bloods, strokes heart attacks…sudden death.
‘The number of out-of-hospital sudden death episodes has increased since COVID-19 outbreaks. One of the possible reasons is the high incidence of major thrombotic events in patients with COVID-19.’
It would therefore seem that caution would be required, if you were to find a way to stimulate the creation of trillions of spike proteins within the human body. Caution.
Anyway, now you know – I hope – why I became so interested in COVID19. Because it links together a whole series of processes that, I believe, are key to understanding cardiovascular disease. Endothelial damage, blood clot formation, the central role of the blood clotting system.
Of course, COVID19 represents an acute vasculitis which comes and goes at some speed and is unlikely to lead to the longer-term damage required to create the repeated clot deposition necessary to drive atherosclerotic plaque formation. However, it can still cause acute clot formation, which can lead to strokes and heart attacks and kidney damage, and suchlike.
It is why, after I got vaccinated, I took aspirin for a month.
Next, fully back to cardiovascular disease – and associated stuff. I will even start to promote my new book – due to launch in October. ‘The enduring mystery of heart disease – The Clot Thickens.’ Yes, it was my son who came up with the title. Not that I will ever let anyone know it was him.
1:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6046589/
2: https://www.sciencedirect.com/science/article/pii/S1074761319300937
3: https://www.sciencedirect.com/science/article/pii/S1074761319300937
4: https://www.frontiersin.org/articles/10.3389/fmed.2018.00200/full
5: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4399148/
6: https://www.nature.com/articles/s41584-020-0474-5
7: https://ccforum.biomedcentral.com/articles/10.1186/s13054-020-03077-0

Excellent job. Good to see you have flexibility in your actions. The alternative is dogma.
Brill!
Great to see you back Doc!
Always relevant, always interesting.
It is why, after I got vaccinated, I took aspirin for a month.
Thank you, Malcolm. I am so pleased you’re up and running again after the events of the last months. Put a big smile on my face to see you’ve resumed your work on heart disease – including what can be learned from Covid.
Ouch! A bit too deep for me Dr K lol, just wanted to say good to have you back and please keep posting and keep making us feel positive it’s getting harder each week just to survive,we can’t pass comments on any websites, we can’t go on holiday ,we can’t visit loved ones because we are not welcome, we can’t go to the theatre.
I must just add I was surprised when you said you had been vaccinated,not a critism just surprised that’s all and good idea about the aspirin,I have read of people taking an antihistamin prior to vaccine too.
Our plan is still to last out avoiding the vaccine as long as we can but the pressure we feel is unbelievable.
Anyway great to hear from you again take care.
while it’s getting difficult, understanding what is going on may help. There is more in this than the link might suggets. https://www.ukcolumn.org/video/doctors-for-covid-ethics-symposium-session-2-the-going-direct-resethttps://www.ukcolumn.org/video/doctors-for-covid-ethics-symposium-session-2-the-going-direct-reset
AhNotepad, that link didn’t work. Not yet more censorship, surely?! Must be worth reading, if so. Interestingly when I had Covid 19 (Dec 2019/Jan 2020) I had chill blains and a rash on my fingers and toes. I don’t know if that is typical of adults. Also, my husband did not have any illness at all so either he was already immune or maybe (like with Ebola and HIV), there are some people who just don’t get it?
The link is broken. It is a partial concatenation of two copies of the URL. The correct link is:
https://www.ukcolumn.org/video/doctors-for-covid-ethics-symposium-session-2-the-going-direct-reset
You can see all the sessions here. The search tool is your friend! 😎
https://www.ukcolumn.org/search?keywords=doctors+for+covid+ethics
Alison, the link was repeated, It should have been https://www.ukcolumn.org/video/doctors-for-covid-ethics-symposium-session-2-the-going-direct-reset
Hi Alison – my husband had the same. His toes were in a bad way for weeks after. Eventually they put themselves right but then, after vaccination it came storming back – on fingers as well, this time. GP not interested.
I wouldn’t simply assume that “it’s too deep for me”, Jeanie. Some very important topics are complicated and look very daunting at first. If you really are interested, you can understand almost anything – Lord Kelvin said so!
The trick is to slow right down. Whenever you come to a word, phrase or idea you don’t understand, admit it freely – and then go and look it up. Wikipedia, pisonous though it can be on anything controversial or political, can be very helpful as its goal is to provide a “way in”. Each Wikipedia article should provide copious links to further reading.
It’s a bit like learning to read a foreign language. Many people have shown that it can be done simply by picking a book you would like to read, in the foreign language, getting a notebook, pen and dcitionary, and just starting. You write down every word or phrase you don’t understand – probably all of them to start with – and look it up. Soon you find that your interest drives you on, and it begins to be easier sometimes to remember a word – or look it up in your notebook – than to go back to the dictionary.
What a fascinating read! Thank you.
Where has the real Dr Kendrick gone? He is now a Covid Clone. Perhaps he has gone to join Flu in the graveyard of tired old clichés. Remember, this deadly virus still overwhelming affects those aged 82.4 with serious underlying health conditions. And you have to be tested to know if you have it. You were right to promise not to talk about Covid .
The good doctor would not have been able to treat, or even see, his patients without getting the jab.
It would seem that being available to them was more important to him. Certainly it must have been a great comfort for those patients, especially if ill with C-19 and even more so if very elderly.
Glad about the aspirin though.
Can’t help saying I agree this was likely his reason. A true doctor caring for his patients, even us who love and follow him who he does not even know. He gave us a somewhat subtle hint to think several times before getting a jab while taking one for the team himself. Similar to the two top FDA vaccine regulatory officials who recently resigned. Thanks so much Doc!
Dr K has said explicitly somewhere that he had to accept the vaccine in order to continue practicing medicine.
When they eventually come for me and hold me down…as I foresee one day. I know myself that I will vomit on the injector.
They wont be coming for a while. They still need enough people not complying so they can stoke the hatred and justify the vaccine passports (or more accurately the digital enslavement ID)
There is no requirement as yet for any UK healthcare professional to have a C19 vaccine. Indeed now that it is apparent that they do not make the vaccinated person less likely to pass it on even if they had it, the arguments about protecting others can go in the skip with much else. https://www.bmj.com/content/372/bmj.n810
Not legally but I surmise that any health organisation that wants can legally introduce such a policy. Also, Dr Kendrick works in a care home, not a hospital, and care workers now are legally obliged to have the RNA jab. (I don’t like to call it a vaccine)
Very interesting Dr. Kendrick, even for a layman following as best he can. “It would therefore seem that caution would be required, if you were to find a way to stimulate the creation of trillions of spike proteins within the human body. ” I’m guessing this refers to the MRNA vaccines. A very good friend, 65 and fit as a fiddle, had a close call (blood clot in his brain and temporary loss of sight) after the Moderna jab. He’s on blood thinners and doing fine. Guessing he won’t be doing the boosters. Not sure they are a good idea for a disease that is likely on its way out. That may be what the situation in Israel is telling us. Time will tell.
Apparently, from a nutritional practitioner’s point of view, those who are fit and healthy and enjoy excercise, are burning off their storage of Vit C so that when they become ill and continue to carry on regardless and not let the infection get them down, do not realise that their exercise is actually depleting the health of their immune system. So it appears that this is possibly the reason why so many people are surprised at how so many athletes/healthy people of all ages are succumbing to Covid 19 and dieing or having serious health problems??????
It is probably NAD+/niacin deficiency that is causing a disproportionate amount of long-haul syndrome in athletes:
https://nkalex.medium.com/the-team-of-front-line-doctors-and-biohackers-who-seem-to-have-solved-long-covid-5f9852f1101d
JDPatten: Have you read Rebecca’s link? Most interesting.
Gary; Rebecca,
Yeah. Niacin, zinc, a few Brazil nuts, etc. Can’t hurt, I suppose. This regimen is never going to get an RCT though: $$!
You only know if your shot in the dark has hit when you turn the RCT light on. Knowledge.
This virus is horrific in more than just causing disease.
Dangerous complacency?
or
Unfounded fears?
or
Feeling lost and confused somewhere between with no Knowledge Entity to truly trust?
Check this out . . . and the comments, to balance out the inordinate decisiveness :
https://www.medpagetoday.com/opinion/marty-makary/94619?xid=nl_marty_2021-09-21&eun=g42415d0r
What’s your rationale? What’s your rationalization? (There is a difference.)
I’m almost breathless galloping through your amazing post, most of which of course went over my head. However I take a keen interest in this kind of research, having had heart problems and rheumatoid arthritis for a number of years. Many thanks for this, lodges in my brain somewhere!!
On Thu, 16 Sep 2021, 09:26 Dr. Malcolm Kendrick, wrote:
> Dr. Malcolm Kendrick posted: ” 16th September 2021 Bridging the gap > between cardiovascular disease and COVID19 [Where two diseases meet] Having > announced that I will not discuss COVID19 anymore, I am about to do so – at > least in part. Yes, you may now be thinking… how can we ” >
Wow.
Thank you.
Forgive me, Malcolm, but I’m going to be very blunt, and even impolite, if not outright insulting.
You wrote an extensive and elaborate analysis and rationale to explain that the only reason you got the jab, is that you also took the antidote alongside it.
Still,
With respect
Martin Levac
I am glad you are a mind-reader. Or maybe not. There was a significantly more important reason for writing the blog. Which a few readers have recognized
I wonder, how much aspirin did you take – the standard 75 mg tablet, or something bigger?
I think 75mg would be fine. This seems the optimal dose for reducing platelet aggregation
Thank you! That is exactly the kind of actionable information I have been seeking. I suspect that if you’re on clopidogrel, that will also help.
I have seen stories that statins reduce severe/fatal covid but I am unsure how much of that is “healthy user bias”.
“Inflammation drives atherosclerosis” seems obvious but good luck getting the average GP to care about it. My GP doesn’t even want to talk about the results of c-reactive protein tests. The only thing he ever says is “take your statin and shut up”.
Thank you for this great, vascular blog, frightening though it was. Thinking (hoping) Nattokinase, Serrapeptase, bromelain, Fish Oil and some other anti-inflammatories such as curcumin, luteolin, black seed oil etc. would also be useful along with the aspirin. If you don’t mind, which vaccine did you choose as was their a particular reason for your choice?
How would the efficacy of aspirin compare to a blood thinner such as Xarelto? I am imagine aspirin would have less risks
How much asprin required`? None at all for the ultimately coerced. Any thinking person would switch a saline solution.
If you remember a few centuries ago, we were debating whether ACE inhibitors were dangerous or not in the COVID crisis. I have wondered for some time if ACE inhibitors (which I still take for my BP) are actually helpful against COVID – ie do they inhibit COVID access to a cell?
Yes, I’ve wondered the same — how does the virus interact with commonly prescribed medications, especially in older people who are taking many drugs.
I suspect you will find that the answer is “No one has the faintest idea”. There are hundreds or thousands of drugs being dispensed by doctors, and hardly anything is known about how they interact. The number of permutations and combinations rapidly becomes hopelessly large – and besides, it’s never easy to do research on drugs that are supposed to be keeping people alive.
Research natural alternatives for high BP : – nitric oxide & vasodilators.
Resveratrol – the black grape compound which may explain the CVD “French paradox” …
If the claims in this paper is true, resveratrol also inhibits the Covid-19 virus too …
Search for : Resveratrol Inhibits HCoV-229E and SARS-CoV-2 Coronavirus Replication In Vitro
URL = pubmed.ncbi.nlm.nih.gov/33672333/
Also : Research vasodilators : Beetroot, pomegranate, Malbec grapes / wine, dark fruits, high cocoa chocolate etc etc
Did I mention resveratrol …
MG
There are also ARBs……angiotensin receptor blockers. I take one for high BP. I’m not sure if they work in the same way to block the ACE receptor from attaching the spike protein. Still investigating.
ACE inhibitors and ARB’s not unsurprisingly seem to cut it. Here’s some brain aching bedtime reading https://heart.bmj.com/content/106/19/1503
I wonder the same. I’m also on anticoagulants which would presumably discourage clots. Sadly that doesn’t include the ones who try to pressurise and guilt-trip everyone to have the jab!
If you read about devastating side effects in some patients after vaccination, and then read that the ADE’s are blockable without reducing the vaccines’ intended protective effect, you will have gained invaluable knowledge. I have been on daily aspirin, and took antihistamines prior to my first jab (to prevent local histamine release) but even as a physician I did not know about this information (thanks Dr. K). I will be much more calm about taking jab 3 armed with aspirin and H1/H2 blockade prior.
aspiration is another factor – i’s not generally done for covid vaccines, unless you ask nicely
A pathologist is reporting 20x increase in cancer cases in 2021–could be delayed screening or vaxx.
Which pathologisr? Where? Give details,give your sources. Unless you are just stirring the mud?
Ryan Cole
https://www.ukcolumn.org/article/stabilising-the-code
Thanks, Sid, that column is a deep dive, but worth it. Links to Ryan Cole’s observation about endometrial cancers being up by an order of magnitude.
Bad luck on the downvotes there, son. Kendrick is an erudite C19 and Cholesterol theory sceptic, but even (especially) he, attracts hordes of sycophantic worried-wellers who will hear no criticism.
Yeah, downvotes and upvotes have a different effect on me than on most. I’m not concerned with the up- or down-. Instead, any vote indicates that the message was conveyed, and was conveyed effectively. It got through and entered the brain, at least enough to cause a response, a primitive response granted, but a response nonetheless.
Indeed, subsequent comments say as much, with echos of “poison no, unless antitode, then poison yes”. I’m not the only one to have gotten precisely that message from Malcolm’s words.
For many, it’s going to take a while before the message gets to those pertinent points of contention in the brain. But the message will get there, eventually. When it does, it’s going to hit pretty hard. I predict variations of “what the hell am I doing?!?”.
With regard to Malcolm and his work, I have the utmost respect for him and his work. In fact, that’s the only reason I would risk offending him. Otherwise there’s no point.
@ Martin Levac / Malcolm Kendrick / Anyone
MK in response to Martin levac wrote : : I am glad you are a mind-reader. Or maybe not. There was a significantly more important reason for writing the blog. Which a few readers have recognized
I am now lost.
I got the aspirin bit.
What have I missed please ?
Why is the message so hidden that it cannot be easily recognised ?
Martin Levac : Did you get a satisfactory response to your statments about aspirin being the reason for the blog post?
Thanks
MG
It is instructive that in order to inform or enlighten nowadays one must resort to codes and hints.
We have come a long way from discussing a pretend pandemic, and I for one have been around the world enough recognise a hard-sell, or a scam, when I see it.
Two years and more in and we’re still banging on about saving the ‘NHS intensive care wards,’ as the Melbourne cops are spraying pepper spray directly into the faces of old ladies on the ground in a head-lock, and the rest of the world are merrily all but holding down 12 year old children for their multiple injections.
As for the coded message, my observation in reply was moderated right out by the Doc so I feel I might have hit the nail right on the head.
Andy, I too never got any message other than ‘take aspirin’, to which I am allergic – as are many others. Could you not try another, simpler code which would get past the censor, for us simpler souls? I suspect you may be exaggerating about the pepper-sprayed old ladies and the forcibly injected 12-year-olds? No? As I say, I am a simple soul who understands plainly-stated truths. If something is true there should be no need for codes, or exaggeration.
You can’t handle the truth. The government (along with Facebook, Twitter, Instagram, the Beeb, Guardian etc.etc.) says so. And so certain people have been instructed, under threat of termination of their jobs, not to tell you the truth. GPs amongst them. Try to keep up with the narrative rather than complaining about receiving hints; the only other option is nothing.
I feel like I’m at one of Kafka’s parties …
MG
A thumbs up wouldn’t do justice. I’m at the same party. Do you like the Hieronymous Bosch murals?
You bad person. I hope the Malcolm groupies don’t find out where you live
OK. Let us move back to a discussion of the issues.
Dr K, (or anyone) I have a question. I read somewhere that only a portion of the spike protein is used in manufacturing “The Jab”. It is claimed that it’s therefore harmless. Is that true?
Thank you so much for all your hard work and sheer stubbornness at refusing to be intimidated by the attacks you get for insisting on following the evidence instead of the party line.
Jeannette as far as I know the coding for the full S-protein is used. (Which is only a part of the virus, perhaps that’s where the confusion is?)
That is, the ‘original’ S-protein as encoded/decoded by the Wuhan lab. Which is now irrelevant in natural transmission. It basically keeps flourishing merely because of the vaccines.
Thank you. After posting the question I came across a paper that said some vaccines use the whole of the spike protein while others use sections of it.
This is fascinating, thank you Dr Kendrick. It makes me think that vitamin C would be extremely helpful, especially high dose intravenous vitamin C in acute cases of COVID-19, and after vaccination. I take 4-5 grams daily, more if I feel a cold or flu coming on.
Good luck with getting answers to that question.
Maybe in around 10 years we will have had enough data and someone has analysed it, but for the time being just think of all those happy shareholders in the drug companies, their deserved good fortune must surely give us all a warm feeling.
Sorry, this was a reply to a question about contraindications to the vaccines.
This is presumably why anti-virals and anti-inflammatory drugs taken in combination at an early stage of the disease are somewhat efficacious. Frontline doctors grasped this very early on and it’s sad to see how they have been crushed by the assorted vested interests.
It’s truly appalling that experts like you have to make readers read between the lines to get across messages about how dangerous the vaccines are, but thanks for doing so anyway.
Here is something that doesn’t require any reading between lines. It’s as clear as it gets, though some will argue otherwise. https://www.bitchute.com/video/e1Q9wrbWlXwz/ It’s Reiner Fuellmich with an update to the current investigations.
Thank you for this, I enjoyed reading it immensely. I will read more thoughtfully as well as the important references you have provided. Will you have the booster vacc? Who is publishing the book? I would pre-order if I knew.
“It would therefore seem that caution would be required, if you were to find a way to stimulate the creation of trillions of spike proteins within the human body. Caution.”
Funnily enough just before receiving your latest missive I’d been reading Public Health England’s “SARS-CoV-2 variants of concern and variants under investigation in England
Technical briefing 20”.
Click to access Technical_Briefing_20.pdf
From table 5 on page 18, there were 13 delta deaths within 28 days of a positive test in the 25,536 fully vaccinated under 50s, or 50.9 deaths per 100,000, compared with 48 deaths in 147,612 unvaccinated, which is 32.5 per 100,000. So the under 50s vaccinated are experiencing 57% more deaths from delta Covid than the unvaccinated. As for the over 50s, the numbers are much worse for the unvaccinated, but no mention of co-morbidities. But for the under 50s, caution indeed.
(Another takeaway is that on page 3 bullet point 4,
“PCR cycle threshold (Ct) values from routinely undertaken tests in England
show that Ct values (and by inference viral load) are similar between individuals
who are unvaccinated and vaccinated.”
Elaborated on page 35:
“This means that whilst vaccination may {or may not! Eggs} reduce an individual’s overall risk of becoming infected, once they are infected there is limited difference in viral load (and Ct values) between those who are vaccinated and unvaccinated. Given they have similar Ct values, this suggests limited difference in infectiousness”)
“From table 5 on page 18, there were 13 delta deaths within 28 days of a positive test in the 25,536 fully vaccinated under 50s, or 50.9 deaths per 100,000, compared with 48 deaths in 147,612 unvaccinated, which is 32.5 per 100,000. So the under 50s vaccinated are experiencing 57% more deaths from delta Covid than the unvaccinated.”
This is not correct because it’s based on hospital admissions which are vaccine-dependent and not the real population cases or infections. Thus your rate of death in the vaccine group is not reflective of the real population because it’s a filtered set and most cases do not go to hospital.
Using just the population vaccination rate, and deaths per cohort – the relative risk of death between unvaccinated and vaccinated is 8-9x greater.
For example, if 1,000 vaccinated people were admitted and 500 died and 500,000 unvaccinated people were admitted and 250,000 died, both death rates are the same by your logic.
Why are hospital admissions vaccine dependent? I would have thought it was symptom dependent. Why would a fewer proportion of symptomatic vaccinated people go to hospital rather than unvaccinated?
Your point on the vaccinated population is valid, and one of the many factors which confound the data, but the review doesn’t tell us the average number of fully vaccinated people in the six months of data.
https://www.itv.com/news/2021-01-06/latest-vaccine-news-follow-the-uk-rollout-of-the-coronavirus-jab-with-our-daily-tracker
gives us some indications of how many people were fully vaccinated in the <50 group at 1st February – approximately zero. That that age group wasn’t even eligible for the vaccine until mid April (except for the vulnerable, who had had their first shot by then), and the whole cohort had received (or at least had the opportunity for) the first jab by July 19th, two weeks before the end of the data set. What is the average? I don’t know, but if you take the ~ 29m over 50s out of the fully jabbed % of 38m at 1st August, it has to be well below 30%.
Nor do these figures take into account non-Covid vaccine deaths. We know from the Pfizer clinical trials that the overall mortality is higher in the vaccine cohort than the saline solution. For AZ the figure was no deaths in either group, but that was a very small, n=2012, study.
Dr Kendrick, I have extensively researched vaccine ADRs over the last 10 years since my daughter had life threatening/changing ADRs to the HPV vaccine. Most of my research in the UK has been by Freedom of Information Act (FOIA) requests since the MHRA, JCVI, PHE, etc, tend provide totally different information to that published publicly. I also research the USA Vaccine Adverse Event Reporting System (VAERS) where studies indicate only 1% of ADRs are reported and Vigiaccess (WHO global database of ADRs) who acknowledge only approximately 10% of ADRs are reported (for drugs, not specifically vaccines). I have just done a quick search on Vigiaccess and found the following amongst the 2 million plus reports of ADRs for Covid-19 vaccine:
Blood and lymphatic system disorders (80227): Lymphadenopathy (62212),
Lymph node pain (8142), Thrombocytopenia (5360), Immune thrombocytopenia (1620),
Lymphadenitis (1610), Anaemia (1294), Coagulopathy (729), Neutropenia (404),Increased tendency to bruise (389), Leukocytosis (380), Leukopenia (300), Lymphopenia (267), Spontaneous haematoma (248), Pancytopenia (232), Disseminated intravascular coagulation (197), Splenomegaly (181), Thrombotic thrombocytopenic purpura (177) …………
Cardiac disorders (94788): Palpitations (32899), Tachycardia (22992), Myocarditis (6223),
Atrial fibrillation (4843), Arrhythmia (4782), Pericarditis (4549), Myocardial infarction (3640),
Angina pectoris (2544), Cardiac arrest (2497), Bradycardia (1953), Acute myocardial infarction (1903), Cardiac failure (1822), Cardiac flutter (1680), Extrasystoles (1430), Cardiovascular disorder (1294), Pericardial effusion (1122), Cardiac disorder (1108), Sinus tachycardia (963), Cardio-respiratory arrest (949), Ventricular extrasystoles (887), Supraventricular tachycardia (615), Cardiac failure congestive (529), Cardiac discomfort (478), Acute coronary syndrome (444), Cardiomegaly (422), Atrial flutter (413), Ventricular tachycardia (346), Cardiac failure acute (309), Cardiogenic shock (300), Ventricular fibrillation (298), Coronary artery disease (251),
Cardiomyopathy (240), Supraventricular extrasystoles (240), Postural orthostatic tachycardia syndrome (226), Atrioventricular block (216), Myocardial ischaemia (208) ……………
But the NHS and UK government still believe what they are told by the MHRA and JCVI ie. the vaccine is SAFE & EFFECTIVE. Are they really so naive or are they actually complicit?
I am not anti-vaxx, just pro-INFORMED choice/consent.
Primum non nocere.
Finally……in parliament:
Predictably, Microsoft Edge blocked the video, but it’s still available on Firefox
Fabulous work! Thank you.
Further to my previous comment, I should have also included the Vigiaccess ADRs listed under ‘Immune system’:
Immune system disorders (27828): Hypersensitivity (12451), Anaphylactic reaction (9094),
Anaphylactic shock (1447), Allergy to vaccine (813), Seasonal allergy (580), Autoimmune disorder (554), Drug hypersensitivity (471), Anaphylactoid reaction (414), Immune system disorder (386), Food allergy (261), Type IV hypersensitivity reaction (212), Type I hypersensitivity (202), Bacille Calmette-Guerin scar reactivation (170), Sarcoidosis (135), Immunodeficiency (117), Haemophagocytic lymphohistiocytosis (104), Immunisation reaction (96), Sensitisation (81), Reaction to excipient (80), Allergic oedema (77), Multiple allergies (75), Immune-mediated adverse reaction (67), Serum sickness (59), Decreased immune responsiveness (54), Allergy to arthropod sting (51), Reaction to preservatives (50), Immunosuppression (45), Type III immune complex mediated reaction (45), Allergy to chemicals (39), Multisystem inflammatory syndrome in children (38), Allergic reaction to excipient (37), Cytokine storm (37).
From personal experience I believe that many of the immune system disorders will take MUCH longer to diagnose.
Brilliantly explained and so obvious once you know. Many thanks.
Yes, DO I not remember the absolute refusal to take an injection a while back. And that they have no hold over him being soon finshed as a Doctor? What happened. He put that sentance separately and stand alone, He knew it was a cave-in. How did that happen do you imagine.
What hit me between my deep, blue eyes was the explantion about the cell membrane. Thank you for the great, yet simple illustration which as usual, was expertly explained! Good, old cholesterol, which holds everything together but which is constantly being destroyed by the wide spread prescription of statins – rather like a dry stone wall, which eventually falls down! How many people with Covid19 have died as a result of being on statins, which have already weakened their vascular system as well as, jeopardising the body’s natural way of repairing itself? My other concern is as to how many people are suffering with conditions such as dimentia and Alzheimers due to the destruction of healthy fat in the brain due to this drug?
Thank you Malcolm Kendrick, for all this thought provoking information, which always leads us on to ask more questions.
Corinna: Excellent point. It would be interesting to compare the IFR for those taking statins to those who are not, but good luck finding any data at all, let alone reliable data.
“They” claim statins are protective from covid.
https://health.ucsd.edu/news/releases/Pages/2020-09-23-statins-reduce-covid-19-severity-likely-by-removing-cholesterol-virus-uses-to-infect.aspx
The researchers found that statin use prior to hospital admission for COVID-19 was associated with a more than 50 percent reduction in risk of developing severe COVID-19, compared to those with COVID-19 but not taking statins. Patients with COVID-19 who were taking statins prior to hospitalization also recovered faster than those not taking the cholesterol-lowering medication.
“We found that statins are not only safe but potentially protective against a severe COVID-19 infection,” said Daniels. “Statins specifically may inhibit SARS-CoV-2 infection through its known anti-inflammatory effects and binding capabilities as that could potentially stop progression of the virus.”
Dexter: Interesting. Like with CVD, the (small? tiny?) benefit appears to accrue from the anti-inflammatory effects.
Interesting point about the statins. A 59-year old friend on statins died of Covid last Christmas – hospitalised with respiratory problems and died of a heart attack. I’m horrified at how many people I know of my age (69) are taking statins. Whenever I’m asked health questions I can see the questioner doesn’t quite believe me when I say I’m on no medications – they then list several of them to check … What have we come to when it’s normal for so many older people to be on statins etc?
read what Sebastian Rushworth has to say about statins here: https://sebastianrushworth.com/2020/07/28/do-statins-save-lives/
Thank you
I am 74. Good health. No meds.
Another great blog, and much appreciated. I am doing my best to avoid increasing the number of spike proteins artificially, as per a previous post. Good luck with the new book. I’ll be ordering one here in Japan.
Thanks muchly. Reliable information is always welcome.Unlike what comes from the “experts” – for example the one I heard saying that the vaccine is 100% effective. Those experts we could do without.
Dr K. Brilliant as ever. Thank you so much for this! In very, very lay-terms (…on the back of a huge amount of reading – mostly well over my head), I had come to a somewhat similar conclusion based around the terrible DIC damage. You filled in ALL the gaps! Anyway, death by ‘Black Pudding’ it is, eh? Not a nice prospect.
Have you read the essay from an embalmer, who has described clots as large as baseballs in COVID cadavers?
Any link to that please?
One of many, thanks to Google
https://www.huffingtonpost.co.uk/entry/embalmer-shares-nightmare-covid-experiences-unlike-anything-ive-seen-before_uk_613f5c81e4b0640100a7ee3e
this? https://www.dailykos.com/stories/2021/9/13/2051954/–The-Clots-Were-The-Size-Of-Pancakes-Texas-Embalmer-Opens-Up-About-Covid-Horrors
Fascinating stuff. Thanks. What, other than aspirin, might help with clots?
John: Eating fatty fish. EPA and DHA (the omega 3 fatty acids) have anti coagulant properties. Good lipids to have in your cell membranes, too.
And if you don’t care for sardines, lamb and mutton also have decent amounts of EPA and DHA.
Research turmeric, ginger, onions / garlic, peppers, Vitamin E.
Also research : nitric oxide & vasodilators.
Resveratrol – the black grape compound which may explain the CVD “French paradox” …
If the claims in this paper is true, resveratrol also inhibits the Covid-19 virus too …
Search for : Resveratrol Inhibits HCoV-229E and SARS-CoV-2 Coronavirus Replication In Vitro
URL = pubmed.ncbi.nlm.nih.gov/33672333/
Also : Research vasodilators : Beetroot, pomegranate, Malbec grapes / wine, dark fruits, high cocoa chocolate etc etc
Did I mention resveratrol …
Galland has a nice protocol within this write-up
https://drgalland.com/coronavirus-protection-protocol/
My understanding is that garlic is s natural blood thinner. Rhubarb has also been mentioned.
I think what I take away from this is to understand what a/the jab does, how it does what before deciding jab or no jab ( booster or no booster). I have already determined that the spike protein is the “thing’ that does the damage – Drs Cole and McCullogh and others concur from their clinical experience – but not with the high level of detail set out above.
I am a simple person but taking “A” to cure “B” to avoid “C” at the expense of inducing “F”, “G” , “H” (and so on) to put you at risk of “C” appears to be the medico/pharmacological equivalent of the “Staircase “illusion. I draw a conclusion after reading ” It would therefore seem that caution would be required, if you were to find a way to stimulate the creation of trillions of spike proteins within the human body. Caution.” but I do not want to be a party to Dr K getting another episode of … ” a knock on the door in the middle of the night” so I will stop there. I think he takes a big enough risk already.
In my own quiet manner, I have asked friends and acquaintances “why would you wish to inject yourself with a “preparation” that induces a non natural production of material that is extensively invasive to your body, causes potentially harmful as well as lethal inflammation to avoid a virus that >99% of you will survive and which can be ameliorated in part by upping vitamins/minerals, in part by moving to – say, Texas – in the hope that if you contract CV19 from any variant of SARS COV2 you can access a suite of early treatments to avoid hospitalisation. I am invariably looked at as a recent visitor from Zog.
Instead of aspirin, which makes me feel ropey, I increased my dose of proteolytic enzymes for a month each time, nattokinase and serrapeptase. I was taking some anyway because I noticed on the doctor’s screen that I had high fibrin which he didn’t mention. My father was invalided and died very early from heart attacks so I and my siblings are very aware.
My biology is quite basic so here is an opportunity to ask what is your opinion? In the hyperbaric oxygen chamber I meet long Covid patients who have all found and use these enzymes. By the way they make good progress.
My brain hurts. Seriously – great article. As usual.
It’s amazing how incredibly complicated the human body and its biochemistry are. I remember about 50 years ago reading Isaac Asimov’s (then up to date) account of the clotting mechanisms. E explained that there were about 30 or more separate interlocking systems, all playing their parts to make sure you neither bleed to death no die of clotting. And, as Dr Kendrick points out, it’s also all harmonised with the immune system. (How not?)
While everyone is entitled to their own opinions about such things, I came to the tentative conclusion some years back that the more I learned about biochemistry the less I could believe that we were created by any kind of intelligent design. We are so complicated – and it all works so well, with fall-backs for the fall-backs – that I can only imagine it all having been perfected by natural selection operating over millions of generations. Just think: in every generation, none of those with any serious defect had offspring. Over and over and over. So almost every living thing you see today is the product of millions of prototyping exercises, culled at every stage by the severest of critics.
Which makes me sceptical of crude attempts to fine-tune the immune system by poking a stick into it.
Well stated.
You could think of evolution as a knock-out tennis tournament with several million rounds – and only one winner.
Or you could think of evolution as fashion. After all, it’s the same old molecules, just arranged differently.
“Dinosaurs are so last-eon. Let’s try mammals instead. I think something in a smooth and hairless might do.”
Spot on Prudence. Our bodies have evolved over 1000’s to combat viruses ans all we need do is support out immune system with sunshine/vitD, zinc, vets A&E. in essence stop eating crap food!
I think if you look at the mathematics then natural selection is dead in the water as an explanation. What are the odds of chance producing just one functional protein?
Quite low. Until you compare them with the periods of time, and the number of “trials” during billions of years. Human minds have difficulty with such enormous numbers.
I think “quite low” isn’t helpful. You might like to listen to a few lectures by James Tour, they’re very enlightening on the numbers. NB Human beings haven’t been around for billions of years.
The answer is1/10 to the 74th. And that’s just one functional protein.
This short video clip of a talk by Alan Watts throws the complexity of the human body into stark relief, from a personal point of view.
Prudence, I really like this talk of his too – think it is quite relevant to today’s goings on
“Which makes me sceptical of crude attempts to fine-tune the immune system by poking a stick into it.”
Brilliant! 😀
Thank you so much. I’ve no medical background whatsoever. However, for years now I’ve started to be curious about how the human body functions, why it thrives, and how it starts to malfunction. It has been a personal journey, and my curiosity only grows about the possibility that there are multifarious ways of understanding illness, that there is a lot of self responsibility involved, that, maybe, despite our lack of medical qualifications, we need to be very curious and proactive in our relationship with our bodies and with our medical professionals. A partnership, maybe. I’ve been reading a bit on the history of medicine (only in Irish context) and its move from quackery to professionalisation, and it has been eye opening. There’s been times during the past year that I’ve wondered, not aloud, if the medical /scientific community are regressing, or if it’s merely what was left of my rose-tinted glasses, have been smashed. Many years ago I heard a highly respected heart surgeon interviewed about health, in general, and if people should interact fairly regularly with the medical professionals for check-ups etc. His response was to keep away from them for as long as possible. I believe he died of a heart attack on the golf course. My own experience to date is that one drug seems to need the addition of another to counteract side effects, having watched aging relatives succumb to this, I want better for myself, as best I can. I’m grateful for the the benifits of modern medicine, during the acute stage in particular. However, when it comes to chronic, my experience, and anecdotal evidence seems to suggest, the medical model only makes things worse.
” I’ve been reading a bit on the history of medicine (only in Irish context) and its move from quackery to professionalisation…”
Doreen, I am (perhaps mischievously) reminded of the story about how Gandhi, on his first visit to London, was asked what he thought of Western civilisation.
“I think it would be a very good idea,” he replied.
Wonderful – thanks for sharing!
Love the book title!
Thanks for another great article. I couldn’t help noticing the subtle way you questioned the logic of vaxxing people with the very thing (or the instructions to make the thing) that causes the problem…
Thank you Malcolm, very interesting article although complicated for my old brain. When you talk about the ACE2 receptors it reminded me of a video I saw last year. It was a doctor trying to make sense of the effects of covid and the fatality rate on older people. He thought maybe a lot of them might have been on medications which increase the ACE2 which in turn made it easier of the virus to take hold? I don’t remember the doctor’s name unfortunately.
The clot thickens.
Toxic exposures of particulates and radiative fields, imbalance, and lack or block of functional nutrition all provide the basis for understanding what is assigned to infection.
The disclosure of a toxic agenda costs disclosure to true accountability, and a negative Economy that has captured and replaced Reason by stealth and guile. or in our normalised or trained ‘thinking’ terms, honesty doesn’t pay our way within a Bandwagon to hell – but grab a mask and jack up some boosters…You’ll be happy and know nothing.
As for the effect of nanoparticulates in blood, particularly graphene, magnifies bio-electric responses in truly novel ways. Sunspots could be much more correlated to heart failure than ever. (Lately data collection of sunspot activity has been changed to give about 30% lower than actual according to ‘suspicious observers’ channel).
Who would have thought that after all the physics of plumbing and all the chemistry of a molecular biology devolved and spun out of the MIC – that’s Military not Medical complex – we should discover the Bio-field, just before it is jammed and managed over by machines of grace and beauty (not!).
Covid the clinical symptoms, is a novel name for a blood disorder, causes yet to be determined.
The shock and shaped charge of its narrative set attention and investment into a redistribution of thought, wealth and power by which to build over the lies of the past as a bright new world.
Yes it is very very dark, but once normalised, a bit less pain, seems like happiness.
Munsch’s painting inside every act of collective compliance.
Biofield research might do worse than look at “the helical heart’ – then listen to Manel Ballister – who used to work in heart transplant surgery field.
Life was designed to be lived by the life that unfolds its function.
Not the mind that seeks to usurp and replicate life.
Live this day well!
Obesity and The Pandemic: New Insights. Has obesity driven not just covid mortality, but the pandemic itself? https://swprs.org/obesity-and-the-pandemic-new-insights/ Very interesting read.
I had been thinking that Covid may be worse in people with metabolic syndrome, a combination of diabetes, high blood pressure (hypertension) and obesity.
If true, this brings up, the nature of foods and eating. One article suggest that hormones have an important part to play – Overeating ‘not the primary cause of obesity’, claim scientists
https://news.sky.com/story/overeating-not-the-primary-cause-of-obesity-claim-scientists-12406990
Leptin, Insulin and Ghrelin could get out of whack and so people keep eating because they don’t feel full.
The problem is that no one is going to fund a study.
The cynic in me suggest that growth (economic) is a paramount driver. So eat more, get sick and then pop more pills and potions. A never ending story of consumption!
Steve,
Take a look at the work of Petro Dobromylskyj (his blog Hyperlipid), or Tucker Goodrich, Chris Knobbe MD, Paul Mason MD, Dave Feldman or Brad Marshall. The are all reading the research regarding the role of seed oils (the ubiquitous cheap-to-make yet high-profit oil in our foods) and all sorts of ill health. (Dr. Knobbe has a few videos showing the rising of diseases in lock-step with rise in seed oil consumption.)
People who are obese are always hungry because they are in fact starving as the excess linoleic acid from seed oils, coupled with excess carbs, has the body storing the extra calories without a way to then release them for fuel, when needed (Brad Marshall’s work regarding Torpor is very insightful on this).
The thread that Dr. Dobromyskyj started to unravel decades ago is gaining many followers worldwide and many new minds are joining the battle.
Re: oils :
See Sally Fallon-Morell – The Oiling of America
For high temperature cooking oil use rice bran oil.
For everything else, including your daily medicine, use certified high polyphenol, EU “Good for you” Drop of Life olive oil :
thegreekoliveestate.com/products/dropoflife
holfordirect.com/drop-of-life-extra-virgin-olive-oil.html
Beware, all EVOs are not the same and all of the shop stuff ain’t worth swallowing. None of it meets the EU “Good for You” polyphenol content. EU Commission Regulation (EU) 432/2012 lists a series of requirements for producers to legally advertise the health benefits of polyphenols in their olive oil.
Unpasteurised butter too.
MG
Re T2DM, Met Syn, etc. I ran across the fact that SARS-COV-2 replicates in our cells using glycolysis (like cancer cells) rather than the much more efficient oxidative phosphorylation (our cells generally) and since they thus only get 2ATP per glucose molecule, they love high levels of sugar. Thus yet another reason why T2DM and specifically hyperglycemia are obvious things we should try to avoid.
Thanks for this Dr.Kendrick
I want aspirin too. Do you recommand 1month after the first shot and continued until 1 month after the second shot ? (Second 1 month after the first dose..).
Thanks
Yes. Although I have no evidence to support this timescale. It just seems reasonable
Excellent. Beyond excellent.
It is wonderful to see you connect the dots.
However, it seems to me that for most of the world clarity and sanity are still a long way off…
I have a few questions…
What is there about aspirin that specifically makes it effective against Covid-19 Associated Coagulation, where CAC differs from DIC? (Maybe I’m wrong, but I assume that the spike protein is the main covid factor in CAC.)
Doesn’t SARS-COV-2 infect the nasopharynx before the lungs to a significant degree? (Surely, some of the virus will make it past all the mucosal surfaces on the way to the lungs, but won’t a large number become trapped in the nasopharynx, which is why it gets swabbed?)
If the nasopharynx were to be infected, wouldn’t virus make it into the lymphatic system rather quickly and from there into the blood? Are we really certain that the damage to the lungs is primarily through direct infection as opposed to damage to the pulmonary vascular system? I offer as evidence “invisible (or “silent” or “happy”) hypoxia,” which seems to indicate vascular damage instead of alveolar damage.
““We didn’t know [how this] was physiologically possible,” says Bela Suki, a BU College of Engineering professor of biomedical engineering and of materials science and engineering and one of the authors of the study. Some coronavirus patients have experienced what some experts have described as levels of blood oxygen that are “incompatible with life.” Disturbingly, Suki says, many of these patients showed little to no signs of abnormalities when they underwent lung scans.”
https://www.bu.edu/articles/2020/3-reasons-why-covid-19-can-cause-silent-hypoxia/
‘Some coronavirus patients have experienced what some experts have described as levels of blood oxygen that are “incompatible with life.”’
Perhaps they should have added “as we understand the matter”.
‘In November 1992 Sir Ranulph Fiennes and Dr Michael Stroud set out to achieve what no one had ever done: to walk and ski – unsupported – the 2700km across the Antarctic, a continent almost devoid of life, in temperatures of -45 degrees centigrade. They walked an average of 35km a day, surviving on what they could carry or haul’.
In one of his books, Dr Stroud states that after returning to civilisation Fiennes and he underwent stringent medical examinations. Among other interesting facts, it turned out that they had exactly zero body fat – which, according to medical theory, meant that they were in fact dead.
Stroud describes how, on a walk across London a an appointment, he found himself on several occasions – I think it was three times – impulsively diving into a hamburger joint and devouring a triple cheeseburger. He couldn’t stop himself – and probably a good thing too.
Stroud has a theory that Captain Scott’s team actually died of starvation. Stroud and Fiennes ate mostly butter on their expedition, because it was the only healthy food that yielded enough calories per pound of weight.
It is these extreme experiences that extend the perimeter of our knowledge.
See also “Did Antarctic explorers starve to death?”
https://www.newscientist.com/article/mg14719961-900-did-antarctic-explorers-starve-to-death/
“Blood samples taken by Stroud and Fiennes during the expedition showed blood glucose levels so low at times that they should have been either unconscious or dying. Yet the two explorers kept going. Stroud concludes that they were metabolising ketones, products of fat breakdown that are used as fuel by hibernating animals”.
Sept. 16, 2021
re the SarsCov2 in/on the mucosal surfaces on the way to the lungs, there is this paper, recently trending at PubMed: “The interferon landscape along the respiratory tract impacts the severity of COVID-19”, by Sposito et al., 2021 A very interesting paper which I am still in process of reading. and digesting.
Thank you.
Brilliant stuff! Thank you for the insights. I’m curious if more cholesterol in the blood would be more protective to your cells to prevent antiphospholipid syndrome?
Very interesting.
Why did you take aspirin for a month after vaccination?
To reduce the risks of blood clotting, I think. Aspirin has that effect, which is one reason why you should take it with caution and in small doses.
I hear some doctors prescribe rat poison and horse dewormer. 😉
Great stuff but my head hurt trying to understand some of it. I think i’ll take an aspirin and kill 2 birds with one stone.
I really like this last blog your ability to lead to a conclusion often without saying it, is inspiring, like your books (I have read 3 of them).
I would like to pose a general question…
I like playing with maths, it’s by background, so I have been following the official ONS figures on Covid mortalities from early on in this pandemic, which suggests a different story from the main stream media.
Just as an example, if you look at these figures in the ONS spreadsheets, by far the biggest predictor of mortality is age, a quick calculation (O level maths !) for the last 12 weeks shows the over 60 age groups account for over 80% of Covid mortalities, and yet this group is supposed to have the highest vaccination rates!
Looking at the lower age groups, the total for the under 40’s account for less than 4% of mortalities, why are we still being told it is the young and largely unvaccinated that are at the highest risk of death and need to get vaccinated or else.
When if ever is the lying going to stop?
“When if ever is the lying going to stop?”
When we are no longer ruled by professional politicians, careerist “advisers”, and business people out to make a quick killing (so to speak).
I wouldn’t hold my breath.
If you want ‘insane’ take a look at what is happening in Australia.
Can I suggest the excellent John Dee s almanac, a Facebook group. He is just publishing his statistical analysis of all things COVID from an undisclosed NHS trust. His job appears to have been to analyse the data without bias to discern what has been happening via the statistical evidence. The findings are eye opening to say the least, and like Malcolm are generally an easy read for the less academic/ mathematical minds!
Thank you. Dr. Kendrick. Fascinating stuff. No wonder the disease and the vaccine provoke a similar pathology.
Thank you isn’t enough…but thank you!
Watch
> https://odysee.com/@Sasquatch:6/Depopulation-By-Any-Means:2?fbclid=IwAR1FSzFJxSKGgNwZEacOZcPdLL8mfwGHlwzbfHj5WAgIdFi4BT49MJ4x9YY
I thought I had posted a comment, but can’t see it, so I’m posting again (I don’t ever comment on anything ever, but this is important to me).
Thank you Dr Kendrick for your work.
Regarding Sepsis, you mentioned it causes the condition known as Disseminated Intravascular Coagulation (DIC), which is the primary cause of death in sepsis. With that said, would it be prudent for survivors of Sepsis to avoid contracting Covid-19 AND avoid the Covid vaccines….?
I hope you, or someone here, can offer me some guidance. Thanks.
I would opine that large daily doses of vitamin C would help to avoid both Covid and the after effects of the vaccines.
Claire, your comment disappeared temporarily – like everyone’s – until moderated, The system here seems to be that you can see your most recent comment – with a warning that it has been accepted only provisionally. If you post one comment, then a second one, the first one will disappear.
Come back a day later, and you’ll see all your comments.
And how interesting the treatment for Lupus is hydroxychloroquine…
Message received and understood – but I have no plans to take the COVID vaccine yet. Happy with my immune system.
And I have had most other vaccines.
I remember early on in the pandemic they were connecting C19 to increased rates of heart attacks and wondering why that was “shocking” news. This article from (I believe) 2017 showed someone was already onto the fact that respiratory illness caused and increased risk.
https://www.ersjournals.com/press/2203/respiratory-infections-trigger-stoke-mycocardial-infarction
I don’t bother to read the Lancet any more, but this, from the PURE study and dairy consumption:
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(18)31812-9/fulltext
My cousin took ill after her 1st Astra Zeneca jab, strange pins and needles from the injection site all the way up to her temples. Took asprin for a few weeks and refused the second jab. Her GP was not remotely convinced it was jab related so where does this leave patients? Take the jab but any damage is never going to be treated as related?
Am glad I’ve been taking D3/K2 for years.
Dear Malcom,
welcome back – I was missing your letters!
May be with COVID – it will take a few years until people start to rewrite the book – like it happened/is happening with smoking, the sugar story, to some extend the cholesterol story or understanding, how “Societies choose to fail or succeed” by J. Diamond (Collapse)
For further motivation, maybe a quote from Mark Twain: „The greatest secret is, to be a genius, and being the only one, who does know it!”
Keep on your great work!
Dr.J.Harter
Von: Dr. Malcolm Kendrick Gesendet: Donnerstag, 16. September 2021 10:25 An: jfharter@aol.com Betreff: [New post] COVID19 and CVD – Bridging the gap
Dr. Malcolm Kendrick posted: ” 16th September 2021 Bridging the gap between cardiovascular disease and COVID19 [Where two diseases meet] Having announced that I will not discuss COVID19 anymore, I am about to do so – at least in part. Yes, you may now be thinking… how can we “
Thank you!!!
Nice article tying it all together. I too was amazed by the role platelets play in the immune response and wrote about it in regards to thrombocytopenia and adenovirus vectors: https://www.geneticlifehacks.com/blood-clots-platelets-and-adenoviruses/. Plus a follow up article explaining the genetic variants involved: https://www.geneticlifehacks.com/adamts13-and-vwf-genetic-variants-blood-clots-and-thrombocytopenia/
This, from Trial Site News, about the jab. Enough to scare the hell out of anyone:
https://trialsitenews.com/covid-vaccine-dystopia-a-manifesto/
Are Hirschorn’s numbers about UK covid deaths among vaccinated and unvaccinated correct?
Yes and no: PHE used their own database, not the ONS, to count the total number of people (both vaccinated and unvaccinated). The PHE database has more people than the ONS (double MD listings, people who left the country etc.), most of the excess are unvaccinated, and therefore they will show a lower ratio of deaths etc.
So yes, scary, but mainly as a case of bad scientific journalism…
Is that why more and more people these days are afflicted with saying : “Yeah no” and “No yeah” ?
Single adverb answers only please …
“most of the excess are unvaccinated”
On what basis do you make this claim?
PHE knows how many people have been vaccinated – but they are less accurate regarding the total number of English people (and therefore the unvaccinated).
For example – among 40-49 year olds in England, 6.4M have been vaccinated – but is that out of a total of 8.1M (PHE) or 7.1M (ONS)? Are there 700K or 1.7M unvaccinated 40-49 year olds? The number of cases per 100K for that age group is 1116 (vaccinated) vs. 880 (PHE total) or 2,130 (ONS total).
Understand?
You still don’t understand. This is the problem with science by ‘press release’. You have to wait to see the paper, see that methods, data and how they reached their conclusions. Then you can compare to other research, what rational they provided for any differences etc. That’s the way it should be. ‘Dr.’ Wakefield has shown us that the peer review system may not be perfect, it’s better than the free for all we have now.
As for the $$$, I don’t know. But t seems nether do you
All that is necessary is that papers and data be published online. Peer review is broken beyond repair.
You should have written that ‘All that is necessary FOR ME is that papers and data be published online’. That’s up to you – just don’t expect other people to have the same standards as you.
Peer review is not perfect, as it depends on other scientists checking the data and conclusions (and going a good job). Not every reviewer has the time or expertise to ask the right questions or identify the problems. This can also depend on the quality of the publication.
We all know of extremely problematic papers that were published in high-quality journals, and it took years before they were withdrawn (such as Wakefeild), only following an extensive investigation. However, most are withdrawn in less time, and many are stopped at the pre-publications stage (145 related to Covid-19 to date).
I would quote Richard Horton, editor-in-chief of The Lancet ‘peer review to the public is portrayed as a quasi-sacred process that helps to make science our most objective truth teller, but we know that the system of peer review is biased, unjust, unaccountable, incomplete, easily fixed, often insulting, usually ignorant, occasionally foolish, and frequently wrong’.
I think it is worse than that. It often simply works to reject anything that disturbs the status quo. I think we should get rid of it completely, let people to publish without it, and allow ‘the public’ to read it, comment on it, and reject it – if required.
In theory, peer-review could be a good thing. In practice it is not.
Wakefield might have been exonerated, (just as his co- author Smith was in the high court) were it not for his professional indemnity insurance not being up to date, unlike p R. Mehra, professor of medicine at Harvard Medical School whose HQ study was retracted by the Lancet and who is subject to much less attention than Wakefield.
Since pharma has captured research, I don’t see much value in peer review as a filter in medical research. It likely keeps out baby and allows bathwater through to benefit pharma. Jessica Rose had a difficult time getting her excellent paper published when it was rejected several times without comment from the reviewers.
At least with online publications we can see the data and do our own filtering and provide comments. Review occurs in the comment section.
I don’t know where your confidence in the ‘public’ comes from. How many people do you think can accurately understand most scientific publications? And how many think that they do, but don’t? I think that we can all agree that most people overestimate their expertise (even experts)…
It could be that the current system is ‘biased, unjust, unaccountable, incomplete, easily fixed, often insulting, usually ignorant, occasionally foolish, and frequently wrong’, but that does remind me of Churchill’s commentary in democracy.
Your proposal may work better in some, more technical fields (that are out of the media spotlight), but I think that the Covid-pandemic has exposed open-source publications as a terrible way to do medical/health research.
By the way – there doesn’t seem to be anything stopping interested parties in publishing their ‘research’, much of it in ‘scientific journals’. And don’t forget the ‘pre-print’ servers that anyone can upload their manuscript to, and many do, before any review (and sometimes even being cited).
Well I agree that the public, in general, would not have a clue as to whether or not any scientific paper is rubbish, or not. I suppose I meant ‘make if available for anyone to review.’ For there are those who are entirely capable of understaniding scientific research. For example, the Lancet paper on Hydroxychloroquine.
‘June 4, 2020 – The online medical journal The Lancet has apologized to readers after retracting a study that said the anti-malarial drug hydroxychloroquine did not help to curb COVID-19 and might cause death in patients.’ https://www.webmd.com/lung/news/20200605/lancet-retracts-hydroxychloroquine-study
The company involved, Surgisphere, just made up the data. Not noticed by the Lancet, but independent researchers realised it was complete rubbish. This was proper review, but had nothing to do with peer-review. It can work, but only if companies release the data. In the case of Roche, and tamiflu, no-one was allowed to see the data used – it was secret. As are all the outcome data on statins, held by the Cholesterol Treatment Triallists Collaboration, in Oxford. When the Cochrane Collaboration asked to review the stain data, they were told it was confidential.
So, yes, for ‘public review’ to work. The raw outcome data need to be released. At present, this simply does not happen.
You may not know it, but much research is known in scientific specialties, but is unpublished. People talk and that’s how a lot of it is spread.
Churchill – the brandy drinking depressive (of the black dog variety) and british empire fanatic who oversaw the bombing of Dresden is well past quoting now.
I guess that should know more than most about publishing junk science than Horton (but he also said ‘I do not regret publishing the original Wakefield paper’, I don’t know if he has learned anything). But Horton also identifies that using a ‘free-for-all’ as a method of regulating scientific publishing is problematic (He quotes O’Connor and Weatherall, saying ”A warning: they stress that anyone who thinks the “marketplace of ideas” will sort fact from fiction is dangerously mistaken’).
Do you think that the ‘marketplace of ideas’ is not even more ‘biased, unjust, unaccountable, incomplete, easily fixed, often insulting, usually ignorant, occasionally foolish’, if not mostly wrong?
It could happen, I suppose. But nothing could be much worse than the current situation. I would certainly give it a go. Having had a number of papers rejected, as lead and co-author without any reasonable explanations. If you want to publish a papers saying that LDL is deadly – no problem. Try getting a paper published saying that LDL is not a causal factor for CVD. Bong! Ironically, the Lancet is probably the most biased journal of them all.
I have the ability to sift data from the “marketplace of ideas.”
Do you think that you can obtain accurate information from research which has been captured by pharma? I’m certain that there’s no bias there, unaccountability, corruption, or hiding of truth there. /sarcasm
Interesting to note that Walker-Smith was actually one of the co-authors to retract the paper (https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(04)15715-2/fulltext) and that Wakefeild was considered responsible to the fraudulent research (https://www.bmj.com/content/342/bmj.c7452.full).
So, probably not a good example
David: There was no fraudulent research. The claim of “fraud” was made by a journalist working for the Murdoch empire named Brian Deer, and repeated by Fiona Godlee, the editor of BMJ, in a BMJ editorial. There were no charges of fraud in the GMC “trial” which stripped
Professor Walker-Smith and Dr. Wakefield of their licenses. The paper, which was retracted entirely to protect Smith-Kline Beecham (now Glaxo Smith Kline). It was a case series of twelve children presenting with autism combined with severe gastro-intestinal dysfunction (and pain). It is now recognized that this is a common affliction, but this paper was groundbreaking, and the retraction set research back by decades. They rightly reported in the paper that the parents of nine of the twelve children reported the regression of their children occurred after the MMR vaccination. This was the shot across the bow that industry and their enablers in government realized required a forceful response. This was their great sin: reporting parents’ concerns about the MMR. I suggest you read Justice Mitting’s stinging rebuke to the GMC in restoring Professor Walker-Smith’s medical license, Also read Dr. Wakefield’s “Callous Disregard.” We are seeing scoundrels of the same ilk at work today on a massive scale, a worldwide scale with the current jabs. Who is to hold them accountable?
Cool ad hominem, bro. Everyone produces bathwater, including you.
Weak minds rely on trusted sources.
As for letting the ‘marketplace of ideas’ deal with bad science, there is evidence that even retraction doesn’t affect the citations (https://gh.bmj.com/content/5/11/e003719.long) – a sort of zombie that can’t be killed. So I’m not optimistic.
I think that we are mainly discussing a specific kind of science that can (to an extent) be understood by most people, and not only experts in a field: nutritional/environmental/medical research based on observational and/or open-source data sources (no one has this same issue with polymer chemistry, geology or quantum physics papers…). Similar to sick people ‘self diagnostics’, lots of people think that they understand what’s going on and can so easily come to the wrong conclusion. This isn’t that easy to people who have years of training and do this for a career.
This kind of data that is very problematic and must be dealt with carefully, and even in the best case will usually not give you a clear answer. This kind of data, and research, should then be connected with other types of investigative techniques (physiological, cellular etc.) to give a better picture and serve as a basis for further research
There was a bunch of fraudulent research in paleontology by one person back in the ’70s and ’80s that was the basis for a lot of other work in paleontology, resulting in huge damage to the paleontological database. Lots of papers and lots of peer reviewers resulting in massive fraud. I think it’s better to let more eyes have a look than trust anonymous reviewers.
Gary, I want to thank you. I don’t think that I could have provided a better summary of the problems with the open publishing than your response – still standing up for a paper where 10 of the 12 authors and the journal published a retraction and stated that, not only have they made an error, but that the main author lied to them. Don’t let the facts stand in your way.
I agree, ‘groundbreaking’ one way to describe this sorry indictment of the UK medical establishment.
Regarding the GMC, this was related to Mr. Wakefield’s conduct as a ‘Doctor’, not his skills as a writer. This was his initial crime – not serving the best interests of his ‘patients’. The evidence of fraud came latter.
As for ‘their great sin’, I expect researcher to research (and report their findings), to put their interests (and feelings) aside, not to report ‘parents’ concerns’. I leave that to journalists. I have a lot of respect for researchers who present results that don’t fit their hypothesis. Do you understand the difference?
As for more research, on what basis? What other research do you have linking Autism to MMR? Come to think of it – what ‘research did Mr. Wakefield actually do? Is this science – or science fiction? Maybe Mr. Wakefield’s greatest contribution is in the field of fraud identification and prevention.
David: It is clear that the etiology of the brain inflammation which results in a child’s regression to autism is complex. Nobody really knows with any certainty what role vaccines, including the MMR, play, but there is no doubt they are a major factor, especially multiple vaccines given simultaneously; timing (age of administration) is a factor, as well. The vaccine court has compensated at least 70 families for vaccine-induced autism. The public does not know the actual number because complainants are forced so sign a non-disclosure agreement to receive compensation for the injury to their child. Autism is new. It has exploded over the last thirty years, since the vaccine schedule has quadrupled. The CDC itself determined that the early administration of MMR (prior to age five) raised the risk of autism by a factor of 2.46 in African-American males. The study team was then ordered to gather in a conference room to destroy the evidence. One of the team eventually became remorseful and spilled the beans:
https://www.vaccine101.ca/post/cdc-whistleblower-dr-william-thompson
In truth, we cannot trust the administrative state, not in the U.S; not in any country. They are unelected; they make their own rules; legislators seem to pay no attention to what they do.
Gary, thank you for continuing to
I agree that ‘the etiology of the brain inflammation which results in a child’s regression to autism is complex’, but at that point I lose you.
Do we have ‘no doubt they are a major factor’? How do you get to that? From one retracted publication, conducted by an electrical engineer? As for ‘The public does not know the actual number because complainants are forced so sign a non-disclosure agreement to receive compensation for the injury to their child’, not true. The number of claims and decisions of the VICE are published (there have been 159 payments linked to MMR+MMRV, out of >146M doses from 2006-2019). Are almost half related to autism? Do you have this data?
Is autism new, or is the diagnosis of autism in new? Do you see the difference?
I was wondering what the safe level of aluminium in the blood was. Aluminium has no purpose in the body. It’s not a trace element, you need absolutely zero atoms. It’s present in very few plants, and not bioavailable to humans when it is. It’s a neurotoxin, implicated in Alzheimer’s, autism, MS, ADEM and others. It gets into our system either by ingesting it in a form that passes through the gut/blood barrier, through using aluminium saucepans, cans, stomach antacids or various processed foods that contain aluminium; breathing it in as welding fumes or fine powders; or injection as an adjuvant in vaccines. It can also pass through the blood/brain barrier.
https://www.sciencedirect.com/science/article/pii/S0946672X17300950
“ When aluminum doses are estimated from Federal Regulatory Code given body weight, exposure from the current vaccine schedule are found to exceed our estimate of a weight-corrected Pediatric Dose Limit. Our calculations show that the levels of aluminum suggested by the currently used limits place infants at risk of acute, repeated, and possibly chronic exposures of toxic levels of aluminum in modern vaccine schedules.”
Mind you, I can’t find a definitive answer. Studies such as
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5651828/
suggest maximum levels in urine, and talk about maximum levels of ingestion, but there’s no clear advice. It’s not really what you pass out, but what remains behind. For mercury, by contrast, which is also a neurotoxin, the maximum level is zero.
So what about aluminium in the brain, where neurotoxicity is most important?
Until recently, there wasn’t really much research. Funding constraints, and lack of brains. Brains seem to be in much demand for research generally, but a group of self-funding persistent buggers at Keele Uni. in the UK have come up with some interesting stuff.
Initially they got hold of five brains from deceased autistic people. Well, I assume they were deceased, the article isn’t specific. But the abstract titillates with:
“The mean (standard deviation) aluminium content across all 5 individuals for each lobe were 3.82(5.42), 2.30(2.00), 2.79(4.05) and 3.82(5.17) μg/g dry wt. for the occipital, frontal, temporal and parietal lobes respectively. These are some of the highest values for aluminium in human brain tissue yet recorded and one has to question why, for example, the aluminium content of the occipital lobeof a 15 year old boy would be 8.74 (11.59) μg/g dry wt.?”
https://www.sciencedirect.com/science/article/pii/S0946672X17308763
Not that 5 autistic brains is much of a sample, but anyway I said they were persistent so they went away and acquired a few more lumps of gray (or white) matter. Every single donor, who all had neuro-degenerative diseases, had high levels of aluminium in one or more parts of the sample.
https://link.springer.com/article/10.1007/s00775-019-01710-0
And as they mentioned in the last link, they need healthy brains as a comparison and had located 21 such samples. In May this year they published:
https://www.nature.com/articles/s41598-020-64734-6
Of the 20 brains (they seem to have lost one. Igor?) they made 191 tissue samples of which only 6 gave readings above 3. Only 2 of the 20 were under 66 years old. Statistically, the results were highly significant. No aluminium, no neurotoxicology. Aluminium = neurotoxicology = brain disease.
The aluminium compounds used as adjuvants with vaccines are very small, easily passing through the blood brain barrier. Nobody knows why adjuvants work to improve the effectiveness of vaccines. Thimoseral, a mercury adjuvant, used to be common, but it’s been almost entirely replaced by aluminium compounds over the last thirty years. In the meantime cases of autism increase.
Note that the ‘placebo’ for the Cervarix (HPV vaccine) was the aluminium adjuvant, not saline. So if the adjuvant is the cause of neurotoxicity, it wouldn’t have shown up in the trials. A similar trick was pulled with the AZ ‘vaccine’, using an immunosuppressant as the ‘placebo’, thus pretty much guaranteeing more infections in the control group.
Eggs, you ask an interesting question – is autism (or numerous other neurological disorders) caused by aluminum?
It does seem that there is some link of neurological disorders to the localized accumulation of aluminum in parts of the brain. Do we know what causes the accumulation? Do we know where the aluminum comes from? Is the accumulation a cause or a symptom? Is this linked to the amount of aluminum in the blood?
How does the changes in diagnostic criteria affect the rise in Autism cases?
I though that autism is caused by mercury/Thimoseral. It’s not?
As for the control group in clinical trials, what do you think the control group should be given? I’ve seen trials using nothing (water), the formulation but without the active components and other vaccines. I can see pros and cons of each. What do you say? What do you recommend?
I recommend reading Chris Exley’s work.
I don’t understand your statement. Are you making an argument for aluminum casing autism, supported by Exley’s publications? If you are, please be more specific.
I recommend you take evening classes in English.
No.
No.
‘No’ what? You’re not making an argument about autism and aluminum? What are you saying, then? Can you be clear?
I can only deduct from your ‘pounding of the table’, by referring to my language, that you don’t have a response to what was said. Can you make an argument based on the science or the facts?
AhNotepad: Indeed, Professor Exley is a real hero in this; that’s why his lab at Keele university was shut down, by the same craven and cowardly academic administrators who know what side the bread is buttered on. His research on autism brains showed Al to be contained within immune cells, whereas in alzheimers brains it was diffused throughout the tissue. The implication in the autism brains is that it was carried there from the injection site.
Actually, I didn’t ask that question. The question should really be, What causes autism? Mercury has been implicated in the past, and has been eliminated from vaccines for whatever reason. Mold’s papers provide convincing evidence, far stronger evidence for autism than smoking for lung cancer, that aluminium does the same. Exposure to cadmium, for example, another known neurotoxin, is minimised as much as possible. Why is aluminium exposure not similarly restricted?
Autism was first described/diagnosed in the early 1940s, the timing of which has been linked to the introduction of some vaccines. But mass, and in many instances mandatory, vaccination against smallpox had already been in place for 150 years. However, thimerosal and aluminium were starting to be used in vaccines (and for other uses) from the 1930s. Plus aluminium pots and pans had been in increasing use since the late nineteenth century. Aludrox is still an OTC antacid preparation (yes, people drink aluminium hydroxide in suspension).
The problem with mercury, aluminium and cadmium as neurotoxins is that, as they have no function in human biology and have very little or no possibility of being ingested in a naturally assimilatable form, they are cumulative, there’s no mechanism for removing them from the blood stream or organs.
Why use a placebo in the control group at all? Can there be a true injectable placebo anyway when just jabbing your arm with a needle can create a reaction?
Eggs ‘n beer: Mercury has not been eliminated from vaccines. It is used as a preservative in multi-dose vials of vaccines, including the flu shot, given to millions of children each year. It is true that the exposure to mercury in U.S. children was greatly reduced after Simpsonwood, but the mercury-preserved vaccines are still produced in vast quantities and shipped to poor countries. And some of the killed-cell vaccines given to U.S. children contain trace amounts of this potent neurotoxin.
If your question is ‘What causes autism’? Good luck. Seems like a giant question with so many factors. I just don’t see evidence for the vaccines being the main factor.
As for Mold’s ‘convincing evidence’, all I see is a correlation, among other correlations presented in other papers, but I don’t see proof of a causality or mechanism. Mold states ‘The presence of aluminium in inflammatory cells in the meninges, vasculature, grey and white matter is a standout observation and could implicate aluminium in the aetiology of ASD.’ and Exley then states ‘the observation that the aluminium content of brain tissue in this group as a whole was significantly higher than the similarly aged control group emphasised the likelihood that brain aluminium content is increased in sAD’. What does that convince you of?
I find it interesting that you see seem to take the autism was’ first being described/diagnosed in the early 1940s’ as meaning that autism didn’t exist beforehand. That seems to be a giant ‘logical’ leap, and I don’t think that there is any basis for that. Not only that, but I would submit that the manor of diagnosing neurological disorders, such as autism, has changed so much over the last few decades, that you can’t just take the number of diagnosis as having any scientific meaning at all.
You ask about control groups. Do you know how a scientific experiment is conducted? A clinical trial?
Looks like this site confirms Hirschorn’s “bad science” using ONS data.
https://theexpose.uk/2021/09/15/30k-people-died-within-21-days-of-having-a-covid-19-vaccine-in-england/
I think that this post confirms that you can make numbers say anything you want, especially if you don’t actually have to prove anything. What does it say?
Can’t you read? Why rely on me?
Oh, I don’t rely on you. At least not for credible information or analysis.
I read the ‘article’ and couldn’t figure out what they were saying – except that it seems like a lot of people have died. They don’t know why, but are insinuating that it’s because of the ‘big bad vaccines’. Is that right? I don’t know, and they don’t know.
But as the good Dr. has taught us – who need proof when you have statistics
Maybe monsters came and got people during the 21 days after the people took the vaccines.
I mean, we know that the vaccines are safe and effective, right?
“Fauci said it, I believe it, that settles it.”
We doan need no steenkeeng autopsies.
You have a very rigid way of thinking. It doesn’t make you stupid, but it limits you.
So, you don’t actually have any support for the ‘deadly vaccines’ theory?
I think that you ‘monster under the bed’ sounds just as good as some other hypotheses’ people though around, there’s just as much proof…
Is it ‘limiting’ to think that, after +18 months, +3B vaccinations, +250M cases and +4M deaths, we should try and base our decisions on real data?
Where are you going to get “real” data? Jacinda Ahern claims to be the only real source of information.
You did a lot of handwaving nonsense raving about “bad” UK data with no analysis.
The Germans and Austrians did some autopsies of vaccinated deaths, showing 30% were due to vaccines.
Where are the US and UK autopsies? Are they hidden in a CIA warehouse somewhere, being classified and all?
You are singularly incurious, which doesn’t speak well for you.
If I understand correctly, your answer is ‘no, I don’t have any data to back up my posts’?
Science without data is just ‘Science Fiction’…
Do you have a link to any publication describing the Germans and Austrians autopsies?
Thanks
“The pathologist however received support from his own ranks, and the Federal Association of German Pathologists stated that more autopsies of vaccinated people who died within a certain time frame after vaccination should be performed.
The head of the “Autopsy Working Group” in this association wanted to make general practitioners and health authorities aware of this. In other words, doctors of the patients who die within a few days or weeks after vaccination should apply for an autopsy in case of doubt or the health authorities should take action.
The Federal Association of Pathologists had already requested this in March in a letter to Health Minister Jens Spahn (CDU), but it went unanswered.”
https://freewestmedia.com/2021/08/03/german-chief-pathologist-sounds-alarm-on-fatal-vaccine-injuries/
You could pre-pay for an autopsy https://bnt-cdn.b-cdn.net/upload/videos/2021/03/zw1gdll2CpLNbcBvDeKm_15_dde3c830879dd49e6b508a6ffc716c48_video_480p_converted.mp4
‘Hand waving’? Did you think that the data was clear and proved the point? Isn’t the whole point of this blog to go beyond the headline – or is that only for show?
I would love to see your data about the autopsies.
Pathology slides…
Re pathology slides
What could be the deciding factors between a person who is protected by the vaccine and a person who dies from the vaccine?
One has the code: ‘Injection Protection CC15-4rFRC’. And the other ‘Death by vaccine injection’ has no present code whatsoever.
Interesting press releases (shame that there’s not actually publications, with figures, data etc.), Schirmacher doesn’t even give an exact number of vaccine-related deaths. Would be interesting to see more data and analysis (age, case-of-death, pre-existing conditions, contaminants, vaccine batch etc.).
With +55M vaccinated people, how many vaccine-induced deaths have there been in Germany, and how many do they think have been missed? Were any of these cases also included in the other publications (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8052499/pdf/main.pdf, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8482743/pdf/414_2021_Article_2706.pdf)?
Are there any other cases of death +6 months after vaccination?
Another interesting point – if lots of these cases are linked to contaminants, that could help lower the rate of serous side effects, including deaths. Don’t you agree?
The autopsies, were a proper sampling of adverse events reports, as ought to be done in the UK and US.
The European vaccine adverse event reporting data is publicly available, isn’t it? And we have a range for expected under-reporting don’t we?
Kind of odd that the article you referenced was published in “Legal Medicine.” Maybe the reason that German pathologists believed that they needed to issue press releases was because of censorship?
Don’t know of any cases 6+ months after vaxx.
Perhaps linking lots to deaths might help, sure.
Did you read about the normal, healthy 15 year old boy who died in his sleep the day after receiving his second jab?
Could be, lots of maybes.
What is a ‘proper sampling of adverse events reports’? What methodology is used? What statistical validity does this have?
As for publishing – there are so many journals published junk, I’m sure they could find one. They are already out there with their findings – what are they hiding? Just seems like a red flag to me.
As for the 15y, no, I didn’t. What was the cause of death? 1 day after vaccinations stands out from the other cases listed by the German pathologists. Can we just say that every person that dies after getting vaccinated, no matter the time or cause, is linked to the vaccine?
Could be, lots of maybes.
Exactly the point. The Corrupt Death Cult (CDC) is not doing their job and issuing reports of investigations of VAERS reports on an experimental vaccine.
Proper sampling–have pathologists look at 40 cases of deaths taken from VAERS and do autopsies. I’d expect that we could extrapolate that with a range of 10% either direction with 95% confidence.
Red flag? Red flag? You only see red flags when they conflict with the official narrative. Jessica Rose couldn’t get a paper published except in Peter McCullough’s journal about vaccine-induced juvenile cardiomyopathy. And you probably won’t see censorship of science as a red flag, either.
“The Sonoma County sheriff’s office report shows a determination was made by a medical examiner that the 15–year-old, who was found dead in his bedroom two days after his second jab, suffered “stress cardiomyopathy” and “coronary artery inflammation.””
If we have ‘Proper sampling’ that we have 16 (range of 14-18) deaths. Is that for all of Germany (~83M) or just for Baden-Württemberg (~11M)? Or are we saying that 40% of suspected deaths are probable vaccine-related (How many suspected deaths are we talking about)?
I think that we all know that the vaccines have side effects, some of them even deadly. The question is how many (or maybe how many are missed), and how many of the reported side effects are actually caused by the vaccine.
You don’t see any red flags here? A paper published in a journal were one of the authors is an editor? Shouldn’t this be published as an editorial?
Did you read the paper? Do you agree with their conclusions? Why didn’t they compare to other countries? Did you notice that they don’t have any funding or conflicts of interest?
Is this your example of censorship, a paper in pre-print? Don’t you think that they, or any of McCullough’s journals, would publish a paper from the pathologists?
No, I don’t see any red flags here. None of these pathologists are making any money from this paper. You are being quite silly here with this emphasis on mole hills and i’s missing dots and some t’s not being crossed.
$$$ talks. No money involved in any of this, prima facie.
McCullough publishes cardiology papers, so myocarditis is up his alley. Not pathology reports unless they involve the heart.
As for the spread of scientific research, I don’t know how it ‘is known in scientific specialties’ without it being published – I don’t know how people just know things. This could be true for unsuccessful research (there is a bias towards successful results in the literature), but I haven’t seen anything like what you claim (especially if the is any funding or investment money involved). In some fields, researchers won’t say anything in public, until they are ready to publish (or patent), if only out of fear.
David RE comment Oct. 11 5:45 am, interesting and thanks for that, I wasn’t aware that this was the case. But most people’s initial impression of Wakefield including my own, probably based on the bete noir taint applied by the med establishment & the established media was that he was a true scoundrel with a mission to destroy all that was good about modern western medicine. And then we find his crime wasn’t really such a terrible thing after all, at least what we civilians would judge as terrible. Maybe you can clarify but was not his crime to meekly suggest more research ?
I think that I read recently that a scientist at one of the US government departments destroyed details of autism after MMR vaccination in ethnic groups; I’m not sure of the actual details as I just thought ‘Andrew Wakefield’ and passed on. He and his colleagues put all the hard copies into a large dustbin, by his account, but he kept copies. Happy to be corrected.
Thanks Penny, that does ring a bell but unfortunately I forget the details.
Penny: Here is Dr. William S. Thompson’s public statement, through his attorney, concerning the scientific fraud the team of researchers were ordered to conduct at CDC:
https://greatergoodmovie.org/news-views/cdc-whistleblower-dr-william-thompsons-public-statement/
Penny, you may have read it recently, but it has been going for years. Brian Hooker had a son who was injected and developed autism of a severe nature. William Thompson was the senior CDC researcher who was involved in destroying the data, but he kept copies, and spoke to Brian Hooker. Thompson was remorseful as Hooker was well known to him. No doubt David will weigh in to villify either or both as trolls or shills or any other insult, and he will continue to support the stories from Brian Deer, who was paid by the Murdock empire, whose son James was a non-exec director of Smith Kline Beecham at the time.
Then there’s the Pluserix vaccine which was withdrawn in Canada (under a nother name Trivirex) https://hitchensblog.mailonsunday.co.uk/2013/04/the-measles-jab-mystery-a-historical-note.html then licensed for use in the UK later that year.
Penny – Correct what? Where would one even start? Can I enter ‘scientist at one of the US government department’ into google?
David, just illustrates your knowledge is not quite as great as you think it is. See my reply to Penny. Then you can get onto doing more research.
Is there any chance that you can publish this information in a scientific journal?
After reading this piece, any thinking person who grasps half of it would not voluntarily consent to being vaccinated. Vaccine mandates are a crime.
In that case, no scientific journal would publish it as their controllers ould prevent it.
“It would therefore seem that caution would be required, if you were to find a way to stimulate the creation of trillions of spike proteins within the human body”.
That would depend on what your objective was.
Every day is a school day on this blog. I love it. Thanks Dr K for the grey matter workouts.
Listen to world sepsis day from 13th of september. Aptahem company.
Yours sincere
Leif Laurin
Den 2021-09-16 kl. 10:24, skrev Dr. Malcolm Kendrick: > WordPress.com > Dr. Malcolm Kendrick posted: ” 16th September 2021 Bridging the gap > between cardiovascular disease and COVID19 [Where two diseases meet] > Having announced that I will not discuss COVID19 anymore, I am about > to do so – at least in part. Yes, you may now be thinking… how can we ” >
Some months back, I watched a YouTube by Dr John Campbell (Nurse Teacher) in which he regretted that the people carrying out the injections were not following the former strict practice of ‘aspirating’ the syringe when giving intramuscular injections. From what I can see of the syringes covered in ‘stickies’ this is probably impossible now, and according to Dr Campbell aspiration to ensure the injection is properly into muscle tissue is no longer thought important in the NHS.
Would Dr Kendrick be able to comment on this as a contributory factor to the vasculature damage that seems to be a feature of all the vaccines?
Thanks, and great to see you putting things together.
You buried the lede but you eventually got there.
So my question would be is it known how long the vaccine will cause you to manufacture trillions of blood clot producing spike proteins? Does each successive shot produce a lessening number or does it get worse? Who knows, right?
Couple this with the completely unknown long term effects and I’m quite fine with my decision to accept the risk posed to me by COVID (not very much at all) and tell the powers that be where they can stick their vaccine.
Here in Australia, like in Canada, US, the non-jabbed are being demonised right from the Prime Minister to state Premiers to the media. We’ve already lost many freedoms and to regain them, we need proof of jabs. One Tasmanian senator said they were coming for us, lock, stock and barrel.
A thoroughly enjoyable read! It’s important to remember this “new” virus was made in a lab. It isn’t found in Nature.
Am I to understand you are suggesting that CVD might be an auto-immune disease?
Mariane. There is NO ‘evidence’ that CV19 was made in a lab, and there probably won’t be any found. However, there IS evidence going back decades of many various patents being granted for various Covid varieties – and the companies/peoples that the patents were issued to are not unfamiliar names !
What is undisputed is that the ‘spike protein generator injection’ WAS made in lab(s). And, incidentally most of these labs have patents previously granted for Covid developments.
Maybe just coincidence ? A bit like it’s coincidental that PHE is based at Porton Down.
The most disconcerting thing about these vaccines is that the Moderna vaccine candidate was sent to The University of North Carolina at Chapel Hill for testing (not trialling but examining for structure contents etc) in November/December 2019 – according to Dr David Martin who exposed all the US Patent details.
Such brass neck on the part of those behind the virus and ‘vaccines’ to come up with a treatment before the Pandemic was declared to require one!
Beautifully clear as always. However the similarities with rheumatic disease complications were noted by me last year, and indeed in May 2020 I suggested to CMO Chris Whitty that rheumatologists should be directly involved in developing Covid-19 treatment. I also (having had no response) suggested to the current President of the British Society for Rheumatology that he should get the Society into the act. Were I President (I was once) I would be screaming for a seat at the table, not least as the way things have unfolded makes it clear that rheumatology expertise in the management of immunological disease would have been invaluable.
This blog merely underlines the point. Action in May 2020 would have saved many lives.
Thanks Malcolm! This is very interesting to me. In late January of 2020, just before covid supposedly arrived on these shores I was in emerg with symptoms of heart attack that took about 3 months to resolve on their own. My son was diagnosed with lupus by process of elimination after being diagnosed with normocytic anemia. He had a positive ANA test (neg. dsDNA). Would you expect a positive ANA test with covid associated anemia? He too spontaneously recovered from his malaise within about 3 months. We are doing everything possible to avoid the vax. But if forced, aspirin will be the antidote! Also taking lots of krill oil for the same reason.
Other half and I had an odd “cold” in late December after he returned from India, where he had attended three weddings. I was particularly annoyed by it as I had only just recovered from a cold, and I don’t get many. Of course, there is no way ever to know if it was C19, or whom I caught it from. However, I bet the virus was in the UK long before January.
Thank you Dr K. I had to read this one very slowly…
All this from the man who claimed that COVID-19 is nothing more than a mild influenza – I wonder…
Please provide evidence that I had ever said anything of the soer
Malcolm,
Although my ability to penetrate medical jargon is lower than most here, I’d
like to take a stab at unravelling what else your post is implying – appart from
the idea of protecting yourself from the side effects of the vaccine by taking
aspirin.
You wrote: “Of course, COVID19 represents an acute vasculitis which comes and
goes at some speed and is unlikely to lead to the longer-term
damage required to create the repeated clot deposition necessary
to drive atherosclerotic plaque formation. However, it can still
cause acute clot formation, which can lead to strokes and heart
attacks and kidney damage, and suchlike.”
By implication, if you take the ‘vaccine’ – perhaps even with frequent boosters – you are distributing little factories for making spike protein all over the body, and that will generate atherslerosis and CVD over time in a large proportion of the population (including sadly yourself) .
Have I decoded your warning?
Why would Dr Kendrick play cute with coded warnings? Fear?
This is amazing and astonishing. I am a lay person at best and really appreciated the way you put this post together. It took some time and thinking but I am now empowered to explain my personal thoughts for my own use of vaccine and/or treatment procedure for Covid-19 when I get it. Greatly appreciate your work! Terrie Sent from Mail for Windows From: Dr. Malcolm KendrickSent: Thursday, September 16, 2021 3:26 AMTo: walker.terrie@gmail.comSubject: [New post] COVID19 and CVD â Bridging the gap Dr. Malcolm Kendrick posted: " 16th September 2021 Bridging the gap between cardiovascular disease and COVID19 [Where two diseases meet] Having announced that I will not discuss COVID19 anymore, I am about to do so â at least in part. Yes, you may now be thinking⦠how can we "
This is balanced and interesting information. Caution is indeed warranted regards such vaccines, considering the mRNA does not remain at the injection site (why would it?). mRNA distribution would also be dependent on administration of the vaccine (aspiration, etc) and maybe if you exercise the shoulder, swim, etc.
I also took 75mg of aspirin a day for a month plus fish oil, turmeric and olive leaf extract. You may also want to take NAC: https://chemrxiv.org/engage/chemrxiv/article-details/60c753ec4c89190f3bad43ca.
A bit too technical for a mere engineer like myself. Just before Covid struck I had the last of 6 coronary stents in 2 years inserted and was offered the new ‘miracle anti-cholesterol injection’. Since this wouldn’t be immediately reversible like stopping taking the statins that gave me joint pain, I thought to do some of my own research and Dr Kendrick popped out of duckduckgo. After much blog reading I declined the offer of the injection, very glad now.
I’ve now joined the dots. 1997, a mild heart attack, put onto aspirin and statins, quickly finding it more convenient to break a tablet into 2 x 150mg rather than 4 x 75mg, ie double the recommended dose. in 2016 a casual conversation with a retired doctor about a scratch that bled profusely led me to reduce the dose back to 75mg. 6 months later a severe chest pain turned out to be a 99% blocked LAD artery. Got this fixed with 5 stents over two procedures, then took clopidogrel in addition to 75mg aspirin for 18 months. 6 months after stopping the clopidogrel chest pains again and another visit for a further stent in the circumflex.
Afterwards I had my light bulb moment, went back to 150mg aspirin and have had two years trouble-free. This is 100% down to reading your blog and I thank you most sincerely.
A couple of weeks ago I was irritated by a piece on Mark Dolan’s show on GBN, the interview with the smug heart specialist trying to sell the wonder injection. Immediately wrote to them dropping your name as a better person to speak to. FF to today when I caught the very end of your chat with Alex Phillips, hope to find it elsewhere to listen to the whole thing.
Great stuff. Thanks again Dr Malcolm
Hi Malcolm,What a wonderful column you wrote today!!! (I’m tiring of telling you that, but you deserve it!) I treated and lectured on the treatment of Periodontal Disease (Periodontitis) for many years before retiring some 10 years ago. At one time the journal STROKE said that Periodontitis was the number one cause of heart disease. Now, whether we believe that or not, this is a very serious disease! And now I know why, and I also know why our treatment recommendation included very little surgery; it was done with the object of eliminating the infection (diagnosed clinically, with laboratory microscopic slides and with cultures); and it required frequent deep irrigations and the use of oral irrigation (the Water Pik and others), it was tremendously successful.I wish I had known the exact thinking you detailed in the article 25 years ago, but I actually cheered out loud when I read it today.Pay attention Doctors!!!!Thank you,Dr. Tom Baldwin22694 Grebe LaneOcean View, DE 19970410-326-6690
Wow! That makes perfect sense. Thank you so much.
Doreen – I agree with you completely.
Posted today – take a look at:
https://www.anhinternational.org/resources/documents/a-blueprint-for-community-based-sustainable-healthcare/
and
https://www.anhinternational.org/who-we-are/
This is from the amazing Alliance for Natural Health. Ahead of their time and so much needed in this Covid climate. We can take responsibility for our health! (and not have to always rely on the drug company’s to heal us (!).
Would it be safe for a person taking Coumadin or other prescribed blood thinner to take aspirin, or, would it be necessary when considering the jab.
‘CCR5 Delta 32 mutation’.
Is that why they call it the Delta scariant? Taking the micturation?
Thank you once again for your valuable insights into COVID-19 and Cardiovascular Disease. Much appreciated.
Lord love you – Dear Dr K – Sense, it all makes sense. What is also exciting are the additional leads that are shaping up – within the nutshell that you have exposed, there is so much meat. Thank you – Breathless on a perch of exploration.
Dr. Kendrick,
I’m glad to hear you’re ok. Also, I think it’s wise that you won’t discuss COVID-19 (that much) anymore. Dr. Rushworth also seems to be avoiding COVID-19 now.
COVID-19 is a largely a political/economic issue and has really nothing to do with science or public health. Here’s one blog I found that shocked me because the blogger (a prominent economist) knew from the beginning that COVID-19 is a Plandemic. Gates/Schwab/Soros and the politicians are behind it (no real surprise). Yes, Gates is a eugenicist and is looking for any excuse to depopulate the world. But the politicians are doing it because the global economy is on the brink of collapse after decades of accumulating massive debts and printing money. Once you reach, negative interest rates, then the only recourse is a Great Reset of the economy…Europe’s pensions were broke as of 2019- hence COVID-19= Great Reset.
https://www.armstrongeconomics.com/world-news/wef/schwab-how-can-one-many-be-so-powerful/
https://www.armstrongeconomics.com/world-news/tyranny/the-stupid-will-believe-it-and-ask-to-be-treated-pandemic-to-depopulate-1981/
https://www.armstrongeconomics.com/world-news/conspiracy/bill-gates-has-been-controlling-who/
I hear you, CK, but I’m not understanding the link between the failing, debt-ridden economy and COVID. How will fewer peasants help the Aristocrats? Why wouldn’t they just let the virus rip through us and claim they can’t help us (if they want fewer pensioners…) Why the need to mirco-manage or daily lives?
I too wonder what the agenda is behind inoculating the global population and I fear this is only the beginning phase of what the elites want to impose. Power is the great aphrodisiac, said
Henry Kissinger
The ten or more articles garnering hatred towards those who do not fully support the neurosis, in the Daily Mail today, proved again to me that there is more to this than meets the eye. Even without looking at insane Australia.
It’s about reducing the cost of pensions. The object isn’t to eliminate the young wage-slaves.
Thank you
I’ve been concerned about my body being instructed to produce spike proteins since I discovered what mRNA vaccines do. Why would I want a vaccine that actually causes my body to produce even a part of the virus. I’ve been trying to find out how much spike protein is produced. Is it the same for all people or does it depend on the person? Does the difference (if there is one) cause some people to react badly to the vaccines and even (as has been happening) to die soon after. What happens to the manufactured spike protein after the immune system kicks in? Is it destroyed? What happens in immune compromised people? Can’t find any info. Just as an aside………..Someone on Facebook made the comment that as we know these vaccines are still in the experimental stage and everyone who gets one is part of the experimental group. Now we all know that properly conducted trials have to have a control group………..so I’m happy to tell people that in not having the vaccine I’m part of the control group. Makes me feel so much better about explaining my refusal..
There is the v@xcontrolgroup.com that you could look at. Concerned scientists have set up a 30 year study.
I will lose my job if I don’t get the shot by November. Thinking of getting the one shot J&J if I have to as it seems the weakest. Wondering if there’s anything I can do to protect myself from the jab? Aspirin is a great idea. Anything else? Lower my stress? Vitamins? Raw salmon? Any ideas welcome.
Your choice appears to depend on the value you place on your job versus the risk you are willing to take with your health by either being injected or catching a virus.
I assume, since you have not so far had any jabs, that you are not considered to be in what are called a high risk group.
My advice to you would therefore be to weigh up the risk of catching this virus and the likelihood of your full and lasting recovery and subsequent lifelong natural immunity against it in the future, versus the unknown risks of accepting experimental, “emergency authorised” only, non-approved, novel gene therapy injections into your body.
To me, it’s a no brainer. Because I trust my immune system over the media …
MG
Thanks MG. I absolutely don’t want or need the government “treatments.” I likely even had the covid last year. I’m 51 and have not visited a doctor for any reason in probably 20 years. I just don’t know how crazy the US is going to get, and maybe I won’t be able to find another job without the shots. I live in a “blue state” and not a “red state.” It’s very stressful. Will the unvaxxed become fugitives? Or will it finally “blow over?” Should I sell my house and buy a RV to travel where it’s “safe” for the unvaxxed and live off the money I make from recycling aluminum cans? Is it Thunderdome time?
I am in the same situation. Best to assume you won’t be able to get another job without a shot. Either move to a Red state or get the shot. Or maybe you’ve got the money to retire already.
It’s not going to blow over. Twenty years after 9/11 and we are still doing stupid, useless security theater in airports – you can bet that 20 years from now we’ll still be doing stupid, useless Covid theater.
Like you I have been thinking of J&J and taking aspirin (which I do anyway) as well as vitamin C, D, K2, magnesium, and zinc. I have a large number of coworkers who got jabbed and none of them died. One of them did get Covid though right after the shot. Which brings us to another point – even if you get the shot, you are still going to get Covid. Prepare yourself accordingly. In particular, lose weight if you are overweight.
Well, you said ANY suggestions.
⚠How to remove a big portion of the Poisonous Covid Vaccine Injection from your body with cupping⚠
NOTE: This works only up to 30minutes after the vaccine.
This cupping method, which a Russian colleague shows here, works only up to 30 minutes after vaccination, in any other case cupping is no longer sufficient … but it is surprising how fast the blood clumps due to vaccination … but it is also clear if you understand the biochemical mechanisms of the functioning of graphene oxides+spike proteins behind it. Again: it is not a virus, it is a bioweapon whose explosive nucleotide is transported via the spike protein using the gene scissors (CRISPR). For better transport, nanolipids are used which overcome the blood-brain barrier via the neurolipin-1 receptor …
file:///var/mobile/Library/SMS/Attachments/41/01/B35DAECB-E458-4A7E-A357-7295074E92DC/telegram_video.mp4
Sorry, try this
https://t.me/endtimesapokalypse/8445
Sorry, that link didn’t seem to work, try this
https://t.me/endtimesapokalypse/8445
Are you a HCW? They can do locums.
Thank you Dr Kendrick 🙂
Dr Kendrick
Two questions:
1) Has the number of CVD deaths increased during the Covid-19 pandemic? I guess some follow up numbers would be available by now.
2) Has the vaccinations increased or decreased the number of CVD deaths?
Very difficult to distinguish between a “CVD death” and a “Covid death” because anyone who dies of CVD (or anything else) who tests positive for Covid is going to be called a Covid death.
Regarding vaccinations, it will be the other way around – anyone who dies of CVD after getting vaccinated will be called a CVD death not a vaccination death. If they die after the first shot, they’ll be called “unvaccinated”.
Better off looking at all cause mortality.
It’s a specific query.
Well Malcolm is firmly back in Baa Baa land but something is clearly trying to pull him out again!! How’s things? Like the old yesterday while I was playing on Fortnite – it plays music (car radio) when you drive a car and it was one of your old songs! Can’t remember any of the words but it was a foreign woman and you used to play her quite a lot. xx
And the point you are making is? Without explanation it is just drivel.
What about taking a good dose of Omega 3 fatty acids ? Here’s a review Omega 3 Fatty Acids and COVID-19: A Comprehensive Review: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7779984/ Would Omega 3 be helpful pre and post vaccination instead of aspirin for those of us who can’t take aspirin ?
Hi dr Kendrick!
First of all I want to express my gratitude for your endeavours to keep the world informed on all things medical. You have helped me a lot with your scientific explanations and I have been following you for years.
Your latest newsletter drew my attention specifically to what you said about you taking aspirine for a month to avoid bloodclotting.
As it so happens I was applying som bandaid on two toes because I had developed some hard patches between my toes which made my foot hurt. And, would you know, those bandaidswcontained the same substance you wanted in your blood to avoid clotting. The manyfacturer cautions even for blood inbalances while using their product. Them I thought of those nicotine and morfine applications through the skin. Why not put some bandaid containing the very substance one wants in ones blood and skip over all the other rubbish in aspirin?
If I sound delusional please break it to me gently because of my advanced age 🤗 because what do I know
Medical error was a large factor in the death of my Mom and my doctor-Dad was misdiagnosed by buddies of his at the celebrated Brigham in Boston, at the end of his life. Plus, the only time I ever ended up in an ER, it was due to medical error. One should always ask how people are self medicating, and what are the possible adverse events of doctor-administered drugs and hospital protocols. It strikes me, from what’s below, that the human immune system has a knee-jerk “get out of here!” response to most of Western Medicine. Probably because, I think, WM has been trying to kill, suppress, or throw the human immune system into utter confusion for going on 2 centuries.
——-
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1993236/
Drug Induced Thrombocytopenia
Hundreds of drugs have been implicated in the pathogenesis of DIT. As noted, DIT disorders can be a consequence of decreased platelet production or accelerated platelet destruction. …
[From Table 1, Drug-induced immunologic thrombocytopenia (DITP): pathogenetic mechanisms:]
Hapten-induced antibody
Drug forms covalent linkage to membrane glycoprotein and acts as a hapten to induce a drug-dependent antibody response
– Penicillin and penicillin derivatives?
Drug-dependent antibody
Drug binds to site on membrane glycoprotein and forms a “compound” epitope or induces a conformational change elsewhere in the molecule for which the antibody is specific. The immunogen can be a drug metabolite
– Quinidine, quinine, NSAIDs, various antibiotics, sedatives, anticonvulsants, many others …
Drug-Induced Autoantibody
During the exposure to a medication, some patients make drug-dependent antibody and drug-independent antibodies (autoantibodies) simultaneously [59;60]. Usually these autoantibodies are transient. On rare occasions, these autoantibodies can persist for a long period of time leading to a chronic autoimmune thrombocytopenic purpura (AITP)…
Finally……in parliament:
Another way of looking at CVD and Covid19.
There is an epidemic of comorbidities (diabetes, cancer, fatty liver, body fat, stroke, arthritis, etc) that are common to CVD and severity of reaction to Covid19 virus.
Suppose that comorbidities are caused primarily by insulin resistance in addition to other contributors such as linoleic acid (PUFA seed oils), mental stress, lead, mercury, pollution, pesticides, herbicides, food additives, pharmaceuticals, etc..
Insulin resistance is basically hyperglycaemia together with hyperinsulinaemia. Result is a dysfunctional coagulation system (platelet activation) in addition to increased inflammation (ROS by mitochondria).
Only viable antidote for comorbidities is a low carb, high fat, adequate protein food intake. This is what I am doing to preserve heart function (28% EF) and associated pulmonary hypertension after MI and stent. Pain from gallbladder/liver issues has ceased. Cardiologist considered me a high risk patient for gallbladder operation since I am not taking any pharmaceuticals (statins, blood thinners, BP meds etc).
What always astonishes and disappoints me is how few doctors offer their patients large fees for furthering their medical education and clearing away their delusions.
I have seen it happen over and over, in some cases with the patient being a distinguished doctor or scientist who just got fed up with being given wrong advice – and did the research him/herself.
Just for 9 hours yesterday I forgot about covid; something that’s not easy to do. Particularly now Dr K has introduced another tranche of scientific analysis to bewilder me. I’m juggling with it but dropping more than I’m catching. (Thank you anyway).
I forgot because an 81-year-old friend went missing yesterday. We meet most mornings about 5.00 AM. I’m walking my dog, he’s on his way to the newsagent to do his paper round. He ‘takes his time’, often walking beside his bike rather than riding it, but he gets the job done. We often have a laugh about this or that, most recently after I’d created a slogan for his work, ‘delivering today’s news tomorrow’.
I was upset that he’d gone missing and went to look closely along the route he takes to ‘work’. It took 9 increasingly worrying hours to find him. Many people searched until he was finally spotted 10 miles from home, confused and dehydrated. Thankfully, he’s recovering in hospital. An infection apparently.
Mercifully I can return to worrying about covid.
You watch, my mate will go and catch something unpleasant in hospital now.
Well done Jeremy for taking the time to care ,its in very short supply these days,thank you for just being a compassionate person that was a lovely read.bless you and I hope he recovers well
Delighted you’re firing on all cylinders and not slumped in a corner somewhere, finally extinguished by those whose minds seem permanently shut down. Really looking forward to getting a copy of “The Clot Thickens” when it comes off the press.
So being on a NOAC, Rivaroxiban will be even better than Aspirin if I get treated?. Why does Reg 174 says I should avoid treatment? Noticed the NHS has a £3b contract for NOACs adverised – si they must be good?
driverjess @aol.com
There’s some noise, but a lot of signal at breitbart and citizen free press.
For instance I read today that over half of the covid cases in Florida are breakthrough infections. Kind of important to know that with the media hammering about the pandemic of the unvaxxed.
Retired NHS statistician John Dee ( a pseudonym) seems to have reached a similar conclusion using data analysis rather than biology. Below is a quote from his Facebook group.
“After spending some serious hours undertaking multinomial regression modelling of 161k emergency department admissions with a raft of covariates to account for confounding factors like age, general health, pandemic phase, bed availability etc I have come to the conclusion that if you really want to see what is going on don’t just count unvaccinated individuals in ICU but go count vaccinated individuals in coronary care units.”
John Dee’s Almanac and Dr Kendrick have been instrumental in enlightening me over the last 18 months to the truth and the lies that are impacting us all. The analysis of data is incredible and shocking.
https://www.facebook.com/groups/johndeealmanac/?ref=share
What is the effect of statins on the cell membranes and in all the patients that died or were severely ill with Sars-Cov-2 how many were taking statins?
Drugs. What do the general public know about them? What we’re told, that’s the answer.
I’ve just read Asheen Malhotra’s latest blog post, which included a good interview with both him and Dr Kendrick.
Inclisiran, the ‘new cholesterol wonder drug’ has a spurious pedigree, in fact it has almost no pedigree. A frightening number of us blindly take stuff like this, believing the marketing blurb. But worse, despite no long-term trials it’s been approved by NICE and will probably be prescribed by our GPs. Nobody can possibly know the long term effects of this drug, good or bad. Presumaby, all we can say now is that those who have been involved with any short-term trials have not actually snuffed it on the spot.
I try and follow sensible advice. Load up on vitamin D, particularly over the winter months (+ a bit of zinc). 75mg Asprirn and a couple of other drugs for my conditions. Long established and trusted drugs I hope. And I exercise like mad (within limitations).
Then I was told vitamin C was generally beneficial so I ordered some and immediately got stuck in. The very first capsule, a thing roughly the size and shape of a rugby ball, got suck in my throat and burst. It dispersed down all sorts of tubes, including lungs. I spent the next 10 minutes trying to catch my breath and the next 3 days coughing up, well, I’m not sure.
Staying healthy is not easy.
Ouch! May I suggest you buy powdered ascorbic acid, which you put into a glass of water? Just sip the water through the day.
Frederica has the right idea! We have been taking between one and ten grams of Vitamin C daily for years, and we gave up on the “horse pills” very early. Vitamin C is incredibly benign, and doesn’t need to be packaged in a capsule or pill.
You can order Vitamin C (aka ascorbic acid) from various stores (including Amazon) by the kilo. Holland and Barrett and other health food shops sell the powder in convenient plastic jars.
About one level teaspoonsful (a largeish teaspoon) is about 3 grams. Annoyingly, it doesn’t dissolve very readily in cold water, so I always take one effervescent 1 gram tablet with my powder – you get the effervescents in those cylindrical metal tubes, available everywhere. But I still have to stir the glass, and eventually after about ten minutes there is no powder lying on the bottom.
It’s best to take your Vitamin C with food – and perhaps your other vitamins and minerals, in case there are synergic effects. If you take more than a few grams, it may also be good to split the dose into two or three taken morning, noon and night. The sign that you have taken too much is a powerful urge to rush to the bathroom, but that shouldn’t happen with less than about 5 grams.
The powder is far cheaper than pills – gram for gram of Vitamin C. Back in about 1975 I used to visit University College London occasionally to fix their PDP11 computers. One day I found myself in a lab where they were doing X-ray crystallography, and while having a coffee with the friendly technicians I noticed big jars of Vitamin C in various places. I asked who had put them there, and I was told the director of the lab provided ascorbic acid free for anyone to take. “Who is he?” I asked, intrigued. “Peter Pauling,” was the reply – the son of double Nobel Prize winner Linus Pauling, who first advocated large doses of Vitamin C.
Nice post Prudence Kitten.
As you said earlier, it’s best to dose vitamin c regularly and evenly throughout the day.
You also mention two other things that resonate here : (1) the great man Linus Pauling, the champion of vitamin c and it’s role in arterial health via collagen synthesis – take note all CVD folk and (2) the DEC PDP11 mini computers took me back to my apprenticeship in the 70s. Hellenia supply their LP5 Linus Pauling blend vitamin c powder just as he prescribed. MG.
Apologies to anyone who was there; they were researchers, not technicians.
You may wish to check the ingredients for your fizzy vitamin C. They probably contain aspartame. I stopped having them when I found this, and that there had been some corrupt moves for Sears to get approval for aspartame after it was rejected in 1973.
Good point, AhNotepad. I am not worried by such small quantities, as I think my diet is good in most ways. But some people might want to avoid aspartame and other artificial sweeteners.
The effervescents are just a convenience – you don’t need them. Adding the powder to warm water would probably help it to dissolve faster.
Dr Kendrick states: “Why is all of this important, you may ask. Because it explains which cells are going to be most damaged by COVID19, and why. Essentially, the cells that are most damaged will be the cells that play a role in the RAAS. They are damaged because they have ACE2 receptors on their membranes. Without this receptor, it is impossible for a cell to be infected by Sars-Cov2, and no damage can occur.”
Therefore, would it be correct to say that taking an ACE-inhibitor (e.g. Lisinopril) should provide a significant level of protection from Covid19 infection/damage?
Or that taking an ACE-inhibitor will leave a lot of ACE2 receptors with nothing to do other than welcoming Covid?
I know this is personal, but given what you know now will you be taking boosters if mandated? It seems these mandates are going to decimate the medical field here in the US, since many, having seen first hand the damage the jabs are causing, are walking away rather than submit to them. Just curious about your stance.
Maybe you want to look at this…
Maybe you want to look at this…
https://www.mdpi.com/2076-3921/10/9/1483/htm
Dr. Kendrick, it must first be said that this is really a fine post with a LOT of stimulating thought and analysis and information. Really excellent.
As is my wont, I will try to follow up on all of what is stimulating and investigate further, not least in some search of more detailed investigation for my own case of “real-time” (increased above normal, which for me is considerable compared to normal) dysfunction of my MSK renal tubules while bound by SARS-CoV-2 virions. 10 days of severe nausea and flank pain.
For instance, was this autoimmune in nature? I would think not — rather, I would think purely metabolic. That is coming from one who has myriad and constant and evergrowing (in number) autoimmunities due to severe PID and overburden of T lymphocytes.
Who knows? Maybe. But autoimmunities can only be terminated by death, and non-replication, of all responsible damaged cells and without continued, chronic fresh damage, in my own unusually long and wide and ongoing experience.
Now for my only challenging question. It has been my (conclusion from) reading of the literature that cytokine storm is a condition not possible except in cases of pretty severe exhaustion of regulatory immune function via depletion of certain leukocytes such as T-reg cells. Cytokines generation, coming from the compromised epithelial tissues themselves predominantly, can persist indefinitely (until death). And many of the larger-in-population, persisting leukocytes can keep on going (in response) in sustaining inflammation while regulation is degraded or nearly eliminated. And this likely includes even the innate lymphocytes, or NKCs, which I have to presume keep on going (rogue, maybe) after adequacy of their regulation by T cells is lost. But they are just as potent in destroying epithelial cells as killer T cells I think. And a much cheaper resource, and hence larger in number — the body (i.e. combination of currently-sensed environmental conditions and evolution) does not waste resources.
And if my hypothesis is correct, we are not evolved to cope with this exhaustion. It is “outside of evolutionary context”. So once it occurs, the patient is in real trouble. But it should be avoidable. And is, I believe. Vit D is one plate of the host-defense armor. Especially with respect to the lower lung tissues. The body does not waste resources, and hence less than optimal vit D in liver results in a lower load of AMPs carried around by the leukocytes and the all-important T lymphocytes (first to detect and first to respond to epithelial tissues viral load). Their available weaponry is less than it could be.
Epithelial tissue health is probably the most important of all — i.e. the nucleus-encoded intracellular host defenses. Degraded by modern degenerative conditions (some aka comorbidities) — big-time. But also by highly advanced age — same thing that causes our ultimate mortality.
Only a limited minority of those infected by CoV-2 (virtually all of us by now, I would think, many times over) will suffer cytokine storm. Why so? This seems to be rarely even commented upon.
I have read and listened to Sucharit Bhakdi, as well as yourself and countless others. Your administration of aspirin seems wise. Bhakdi is really highly critical of the new “gene” vaccines, and considers them excessively dangerous for most.
I certainly agree, but biological systems (and earth, as a stable system supportive of long-term cellular life) are chalk full of negative feedbacks. Nothing but. Positive feedbacks are unstable, and hence selected out by evolution. Rather quickly, I would think.
Hence, it is just amazing how well our immune systems cope with alien insults such as these new huge-molecule NA vaccines, in general. The analogs of this type of insult have been present, and are encoded/adapted for, within evolutionary context even though these new-tech vaccines are doubtless without. But nevertheless, death by vaccination seems to be 10x to 100x (or possibly more) than for conventional vaccines such as flu shot based upon the anecdotal and otherwise suppressed and muddied record so far.
Bhakdi is right, but I would just be a bit more optimistic based upon all of the unimaginably complex set of negative feedbacks apparently preventing catastrophe in most cases of artificial innoculation.
Any comment?
May I bring to everyone’s attention the latest superb (though long) article by Simon Elmer:
“The UK ‘Vaccination’ Programme. Part 1: Adverse Drug Reactions and Deaths”
https://architectsforsocialhousing.co.uk/2021/09/15/the-uk-vaccination-programme-part-1-adverse-drug-reactions-and-deaths/
Don’t be put off by the site’s title “Architects for Social Housing”; that is Mr Elmer’s main focus. Although he posts rarely on Covid-19, his articles are absolutely marvellous – deeply researched, absolutely logical, and very instructive indeed.
To deal with the unlawful requirement for care workers to submit to accepting an experimental arm spear (x2)
https://www.thebernician.net/pubs-three-notice-process-to-stop-care-homes-mandating-vaxxes/
may be helpful.
Brian, I’ve read all of the books by this UK family practitioner. This article is interesting. Van
Van Valenta
>
Thank you again Malcolm, this is amazing and wonderful biochemical refresher course for me. Extremely relevant, as the FDA has just voted 16-2 against booster shots, citing cardiovascular deaths from the vaccination.
https://theexpose.uk/2021/09/18/fda-experts-reveal-the-covid-19-vaccines-are-killing-2-people-for-every-1-life-saved/
Robpankratz: It is a near certainty that FDA will overrule them. This is, after all, an advisory panel with no authority. Francis Collins (head of NIH) has already publicly stated that this will be the case, and Fauci just informed us they are hurrying an approval for the poison death shot for the 5-11 age group. These people are simply evil, pure evil, with no redeeming qualities whatsoever.
Even if overruled, the panel vote could be of use in a RICO case against the FDA and pharma.
Dr Kendrick,
Wonderful post which will reward careful reading from even the medically semi-literate (I like to think of myself that way). Thank you for going to so much trouble with this one. And, yes, it is exciting to see one’s dots connecting up.
“Not that I will ever let anyone know it was him.” (We won’t tell a soul!)
This is much like watching “Andromeda Strain” in the theater. Scary, even though fiction.
Don’t want to leave your comfy seat and go out into the differently imagined world.
This, though, however much we the people make “theater” of it, is real.
All the more reason to not want to leave the comfy blog and go out into the real world.
However, cardiovascular disease, per se, is just as real as COVID, and has claimed many more victims. Probably will continue to –– unless we (or someone, anyone) can get on top of it.
It’s what this blog had been all about.
JDPatten: Indeed, and it has lowered the risk for all of us who have taken this knowledge to heart. For me the greatest of these truths is the understanding that how we cope with stress is a crucial factor in protecting us from CVD. What has been the greatest revelation for me about coping with stress? Forgiveness. Forgiving myself first of all, for being such a moron at times, and then forgiving others.
Hello, Goran.
Are you still with us? Been a long time.
Anyone Know??
Hmmm – I was wondering about Goran as well!
Dr. Kendrick’s last citation ( #7), ends with a list of Abbreviations. Some of us find it helpful to print these to print these out although you will want more more…..
Sept. 19, 2021
Sorry for the incoherence above. I was interrupted while writing.
It may be helpful, while reading the papers that Dr. K lists at the end of his blog post above, to print out for reference, the list of Abbreviations. which appears at the end of the last (#7) of Dr. Kendrick’s cited papers. However, in the papers he lists, there are additional abbreviations, some of which should be understood, in order to begin to try to understand the papers. In short, there are lots of abbreviations. StatPearls, found via PubMed, may also be helpful. There , one finds, for example, a long list of interleukins (the ILs) which are players in the immune, inflammation, and coagulation systems.
Thank you to all authors who are aware of the problem of ubiquitous often over-abundant abbreviations, and make efforts to address this problem for the common reader.
So the British Heart Foundation publishes a study from Bristol University showing that the spike protein causes inflammation in the small vessels of the heart….and it gets pulled from their website. Not at all suspicious!
Fortunately – thanks to Wayback – it can still be viewed here:
https://web.archive.org/web/20210913081214/https://www.bhf.org.uk/what-we-do/news-from-the-bhf/news-archive/2021/august/covid-19-spike-protein-binds-to-and-changes-cells-in-the-heart
Helen,
Yes, I checked. The article is no longer there. It seems that any information to suggest that spike proteins can cause damage are getting censored. The article itself states the following:
Covid-19 spike protein binds to and changes cells in the heart
28 August 2021 Jennifer Mitchell
The spike protein found on the surface of Covid-19 virus cells causes changes to cells in the small blood vessels of the heart, according to research we funded presented at the European Society of Cardiology Congress.
Researchers from the University of Bristol have found that the spike protein binds to cells called pericytes which line the small vessels of the heart. This binding triggers a cascade of changes which disrupt normal cell function, and lead to the release of chemicals that cause inflammation. This happened even when the protein was no-longer attached to the virus.
There is some previous evidence to suggest that the spike protein can remain in the blood stream after the virus has gone and travel far from the site of infection. In this study, researchers only studied pericytes from the small blood vessels within the heart. However, pericytes are found within small blood vessels all over the body, including in the brain and central nervous system. This latest finding may start to help explain the effect of the virus on organs away from the site of the Covid-19 infection.
Researchers took small vessel cells from the heart and exposed them to the spike protein. They found that the spike protein alone was enough to disrupt normal cell function, and lead to the release of chemicals that cause inflammation.
They then blocked the CD147 receptor and found that this prevented the spike protein from causing some of the changes to the cells. However, the inflammation continued. Now the researchers hope to find out if a drug blocking CD147 in humans can help to protect people from some of the complications arising from Covid-19.
Professor James Leiper, our Associate Medical Director, said:
“Covid-19 has presented an unprecedented challenge for the cardiovascular research community. There is still a lot that is unknown relating to how the virus can impact our health in the long term, but this research brings us one step closer to better understanding how Covid-19 affects the heart and circulatory system and may ultimately lead to new ways to protect the hear
Why does an academic paper get censored?
Because it presents conclusions and supporting information that powerful people would prefer to suppress. It’s hard enough now to get people to line up docilely for their “jabs”. Every time they hear about the spike protein which will ravage their blood vessels, I expect they will be a lot less willing to have it pumped into them.
It will be worse. Even if people don’t get jabbed, unless you actively take care of your immune system they are at increasing risk because of breakthrough cases. It’s why mass medical treatment (jabs and drugs) of any sort wrecks the system, and increases the problems. (In the case of this fraud plandemic that was the intention).
https://www.ukcolumn.org/community/forums/topic/why-is-there-a-correlation-between-the-vaccine-roll-out-and-increased-covid-19-m/page/42/#post-170868 This contains a couple of links about the 251 times viral load the vaccinated can carry if they get infected later.
Did you even read the whole press release? The authors conclusions seem to be to go get vaccinated.
AhNotepad – Did you read (or understand) the link you post? Did they actually read the paper (still a pre-print)?
Any chance that you read the actual paper? Do you know what they tested or what the results are? What the results mean?
I think that we all know that vaccinated people can get infected, and some can even get sick (and that the AZ vaccine is less effective at preventing infection). When vaccinated, people get infected (and especially have symptoms, as all the subjects did), the viral load seems to be similar.
This paper shows how dealing with Delta is a whole different story than previous strains, with higher viral loads (that’s why they recommend keeping masks etc.). They didn’t compare the CL to unvaccinated people (infected with Delta).
FYI – all the subjects got well, even though the CFR in Vietnam is 2.5%.
As for the second paper, here the transition was from a person vaccinated with Covaxin (an inactivated virus-based vaccine). We know that even vaccinated can get sick (the rest had mRNA-based), but it doesn’t show that they transmitted on.
David, that’s bullsh!t. Vaccines provide immunity. That’s been the definition for 225 years since Jenner. These shots don’t provide immunity or, if they do, it’s for such a limited time as to be irrelevant. So they aren’t vaccines.
Are we talking science or linguistics?
You can call it what ever you want, it’s just semantics. I understand that you are going to take one of the inactivated virus or subunit vaccines? Is Webster OK with those?
If you want to have a scientific discussion, then show me data. If you want to have a political discussion, then you don’t need data – do you?
By the way, I’m still waiting for more on your story of widespread deaths in nursing homes down under. Did they solve the problem?
It’s neither science nor linguistics. It’s lying. They are not vaccines, therefore injecting people with them doesn’t make the person vaccinated.
Nope, nothing to report from the aged care facility. They’re all still dead. Although judging by the number of people lurching around with masks on some of them may have become zombies. Either that or it’s hypoxia.
Here’s some data.
In the US in 2019, there were 123m people aged 18-49.
https://www.census.gov/data/tables/2019/demo/age-and-sex/2019-age-sex-composition.html
For the duration of Covid to September 11th there were 59,527 people hospitalised in that age group.
https://gis.cdc.gov/grasp/COVIDNet/COVID19_5.html
So your chance of catching Covid and being sick enough to be hospitalised over the 18 month period is 0.048%. 1 in 2,000. Or, over a two month period, 1 in 18,000. But, this is hospitalised with Covid, not because of Covid.
Cases are identified by reviewing hospital, laboratory, and admission databases and infection control logs for patients hospitalized with a documented positive SARS-CoV-2 test.
So if you’d torn a cartilage, and tested +ve at hospital, you’d be in the stats, regardless of symptomatology.
Moving on to the risks associated with the ’vaccine’. Here are the statistics for the six month clinical trial for Pfizer.
https://www.medrxiv.org/content/medrxiv/early/2021/07/28/2021.07.28.21261159/DC1/embed/media-1.pdf?download=true
Table S3 | Participants Reporting at Least 1 Adverse Event from Dose 1 to 1 Month After Dose 2 During the Blinded Follow-up Period is on p.11. Trying to equate the risk of hospitalisation from the ’vaccine’ with that of Covid, serious adverse events were defined as any untoward medical occurrence that resulted in death, was life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, or resulted in persistent disability/incapacity. I suspect that any persistent disability/incapacities that didn’t require outpatient treatment would be outnumbered by the ‘with Covid not because of Covid’ numbers, and is this a reasonable proxy for comparison with Covid hospitalisations.
So the excess risk of being hospitalised by the ‘vaccine’ is 0.51%, or 1 in 200, over a two month period, compared to 1 in 18,000 over an two month timeframe if you catch Covid. So for the under ‘50s, ‘vaccination’ carries a 9,000% increase in risk of hospitalisation than catching the bug.
P.12 of the Pfizer trial supplementary shows OM slightly greater in the ‘vaccine’ group, after six months. So it cannot be claimed that it saves lives.
Doesn’t save lives. Doesn’t provide lasting immunity. Doesn’t prevent infection. Doesn’t prevent the spread. Vastly increases the risk of hospitalisation.
It’s not a vaccine. It’s actually much worse than useless. primum non nocere!
By the way – the papers didn’t evaluate unvaccinated people, or compare vaccinated to unvaccinated, so you can’t come to a conclusion regarding immunity based on either publication, can you?
First, I think that we can all agree that definitions have changed, with the science, over the years. We don’t keep everything from 225 years ago. If that was the only conditions, lots of people still wouldn’t be voting.
I don’t think that we should consider a dictionary for scientific definitions. Even Robert Malone calls them vaccines
Second, as for your ‘Vaccines provide immunity’, what is your threshold for immunity? Does the flue vaccine provide ‘enough’ immunity? What about the polio vaccines, or should we only consider the attenuated? What about rabies? Is it still a vaccine if we administer it after the bite has happened or not?
Don’t let yourself be limited by your vocabulary.
The Charities associated with many medical issues are no longer ‘the independent supporting bodies’ that caring earnest people once started. They ran on a shoestring at first but once the ‘magazine’ and then ‘the mailing’ and ‘the advisory help line’ became supported with donations from the pharma’ industry …and who can blame these nice folks for accepting the money ….they become part of the pharma industry and so difficult findings such as Bristol UNi’s paper will be censored and removed. They have been bought.
andy, it’s worse than that. Much worse. These so-called charities are now funding the pharmaceuticals. Where do you think the money the Heart Foundation collects for ‘medical research’ ends up? Or the Cancer Foundation? MS Society? etc. etc. And having used your donations to develop and market the latest statin or whatever, do you ever receive a percentage of the billions of $$$ in profits they reap?
I found the article.
https://www.bhf.org.uk/what-we-do/news-from-the-bhf/news-archive/2021/august/covid-19-spike-protein-binds-to-and-changes-cells-in-the-heart
They implicate the spike protein as causing damage to the heart, then say vaccination is a safe and effective way to avoid this. Vaccination with a vaccine which causes the body to MAKE the spike protein! WTF!
The British Heart Foundation has always bad-mouthed Cholesterol. It specialises in simplistic nonsense. Someone probably pointed out to them that the vaccine itself created spike proteins and they panicked. They are allowed to blame Covid but not the vaccine.
Don’t forget that this is only one part of the picture. There are lots of questions left, some also raised in the press release:
Does this also happen in vivo (in the human body)? We know that not every thing translates from the lab to the whole organism.
How does this differ from the vaccine(s), and do different vaccines behave differently?
How does this piece of information fit in with the other things we know about the COV-SARS-2 and the hart?
I know that we all like to leap to conclusions, but still
Spike proteins will affect the heart. This is true whether you get the spike proteins from Covid itself or from the Covid vaccine. Everyone is going to get Covid eventually, and therefore everyone will be exposed to spike proteins eventually. You cannot avoid spike proteins by refusing the jab. So the dilemma is – are you better off being exposed to spike proteins through the vaccine or through getting the disease? This could depend on such factors as your age, obesity, and general health.
I would really like to see a study of the effect of vaccines on people with a pre-existing heart condition, but all I see is hand-waving rah-rah “of course you should get vaccinated it’s safe and effective” talk.
Dexter
As I understand it, courtesy of people like microbiologist Dr Sucharit Bhakdi, when we are infected with Covid its spike protein is dealt with in the respiratory areas of the body by the immune system. But when you take the shot in the arm there is the risk that cells all over the body will be taught to MAKE the spike protein and thereafter be attacked by the immune system, with potentially a huge amount of inflammation and clotting. So it would seem to be a matter of quantity and placement. I personally think it better to get Covid naturally whatever one’s circumstances.
Adding to Tish’s comments, it’ll be like the other coronaviruses. You’re constantly exposed to them, and your immune system is constantly fighting them with varying degrees of success. I haven’t had a cold for years, the occasional sniffle for an hour or so, or the flu (not a
coronavirus) for 40 years. So I feel fairly confident that I fall in the 81% with cross-reactive immunity, the virus will be dealt with starting as soon as it touches my skin or mucous membranes and won’t get as far as spiking my heart.
https://www.researchsquare.com/article/rs-35331/v1
The key thing is to keep your immune system active. Not just prepared, as with C, D, K, eggs, beer etc. but active! Constant exposure to acute disease is essential for an effective defence. All these measures taken to prevent infections will come back to bite people really hard. Forcing vaccinations on teachers? Unnecessary. The kids’ bugs are providing them with immune system exercises.
https://www.bmj.com/content/374/bmj.n2060
No one asks us in the funeral industry. “No busier than we ever have been,” is the answer.
For quite a while now, I’ve been considering western medicine, and it’s focus on symptoms and specialisation of any one area of health. Specialisation may indeed be needed, but it will tend to make innovative connections less likely.
For example, this came up in one of my searches and says “Rheumatoid arthritis: New ‘highly effective’ treatment on the way after scientific breakthrough https://inews.co.uk/news/science/rheumatoid-arthritis-new-highly-effective-treatment-breakthrough-1185281
Another thought, then took me here “Ancient Remedy a Promising Cure for Clostridium Difficile”
https://research.umn.edu/inquiry/post/ancient-remedy-promising-cure-clostridium-difficile
In yet another example, a neurologist has suggested that c diff may be the cause of Alzheimer’s!
I’m not a doctor, but perhaps me not being a doctor allows one to think outside the box?
There seems to be a debate around germ theory and terrain theory at the moment. Whilst many of these discussions seem to be polarised, I wonder whether it’s about time they were considered together?
Be interested in your feedback, Dr Kendrick (if you have the time)!
Indeed, just like in the ‘fifties a cure for MS was ‘just around the corner’. And in the seventies interferon would eliminate all cancers. Leaky guts, and the solution (diet, non-inflammatory foods) have been known about for decades, it’s only new to the blinkered conventional medics. The same diet eliminates other forms of inflammation too, including those leading to heart disease.
I had to laugh at this:
https://www.newsamericasnow.com/latin-america-new-could-this-snake-hold-the-cure-to-covid/
a) the answer is no, it’s not a cure for Covid. Ebola, possibly.
b) homeopaths have been using this for 170 years.
c) arsenic is the closest homeopathic remedy for Covid. It’s been used successfully in India (Kerala state) and Portugal. However one can imagine problems in marketing arsenic as a cure for anything.
Totally illogical when you consider the toxicity of chemo for cancers, or warfarin, or………spiked proteins 👹
Hello Dr. Kendrick, it was really good to see you and Dr. Malhotra on GB news talking about your favourite topic of cholesterol-lowering offerings from Big Pharma. It is interesting to see that at least one mainstream media outlet will allow an alternative view. Let`s hope that eventually one of the MSM will start to allow alternative and compelling views on all things to do with Covid.
More on pulmonary artery hypertension. Either I can do something to stop the progression or look forward to a lung/heart transplant.
It appears that the initial stage is caused by smooth muscle cell proliferation and migration under the influence of hyperinsulinaemia and hyperglycaemia. This creates a hypoxic condition attracting macrophages, B cells, T cells, neutrophils and a multitude of inflammatory molecules that clog up the arteries. Platelet activation will result in blood clots. This is a different situation than in large arteries with vasavasorum.
The solution is quite clear: reduce glucose and insulin spikes. Also inducing autophagy to reduce misfolded proteins and other crud will increase cell vitality. Autophagy might also clear spike proteins from cells introduced by the Covid19 vaccine.
Having held off for a few days before wading in to this fearing some impenetrable medical jungle, i laughed out (a little) loud – when i came across some neat stepping stones –
“In super-short version:
Infection → inflammation + coagulation → (if regularly repeated) atherosclerotic plaques = cardiovascular disease”
Spoiled we are –
Wow, that was quite a read! Complicated stuff made readable and (almost!) understandable.
Thanks
It would seem in Lithuania now since September 12th it’s a choice of ravaging your blood vessels, or travel, food and life. Coming to everyone soon, spike proteins or not.
Dr. Kendrick,
What do you think of the 50 ng/ml serum vit. D guideline for optimal immune health that Robin Whittle has advocated?
And I’m also curious whether you think that calcifediol should be trialed as regards hospital treatment for acute viral infections. Or would you expect it to be better in outpatient use?
I think speaking to David Grimes would be the route here.
Thanks. From Grimes’ blog,
“The use of Calcifediol is more physiological, optimising circulating blood levels and provide a source of the precursor of Calcitriol to be produced within the immune cells. Normally we would not expect Calcitriol from the circulation to become active within immune cells where so much of it can be produced.
In practical terms it would seem to be the best plan for clinical doctors to prescribe Calcitriol 0.5 micrograms daily for 14 days in the treatment of patients with Covid-19 pneumonia, until our national leaders approve of Calcifediol to be used in appropriate dose (already worked out) in the treatment of human beings, in addition to cattle.”
http://www.drdavidgrimes.com/
Did doctors go through grueling years of study in medical school just to have their hands tied by incompetent/corrupt overlords?
Scroll down to Dr G’s blog of 11 Sept. 21 ‘Covid-19 & Vitamin D – How do we get out of this mess?’ Scene – 10 Downing Street Prime Minister’s Office. It might be parody / satire of a fictional conversation between the PM Bozo and Tweedledee (Whitless) & Tweedledum (the one with £100k’s in vaccine makers). It is believable that this conversation could have taken place.
Aside – I have my Mum taking 8,000 IUs of Vitamin D (& push for more in the winter). I try and make her go out in the sun but she is reluctant. A recent test blood test had her Vit D (OH 25) at 180 nmol/L which I was over the moon with. I get my Dad taking similar & he will go out and sit in the sun plus plays golf.
Dad is pretty rubbish at social distancing (mainly being hard of hearing so he ‘needs’ to get up close to hear). He went down with a lurgi last Friday, followed by Mum Sat/Sun then me, who look after them, Monday evening. From the symptoms, I believe this has been a flu not covid. Short story is that both in their 80’s have soon batted it off – not back to 100%, but not far off. I don’t supplement with Vit D as I soak up as much sun as I can during the summer but a test from work, taken at the end of March 21, revealed that, from January onwards, I should start some supplementation until the sun comes strong enough again from April (UK).
An update for anyone interested. The amount of advice that my parents have had regarding that they should have been jabbed – Mum’s cousins wife (a friend) told Mum that she told her that she should have been jabbed then describes how her doubled jabbed daughter-in-law has caught covid (again) and is pretty bad (as I bang my head against wall). The scary thing is that I now believe Mum is being so coerced, from all angles, including my brother, that she is going to succumb. A further aside, Mum spoke to a member of the dance club (as she has missed the pass 2 or 3 weeks) and it seems that all the double (& now triple) jabbed have gone down with coronavirus (including one hospitalised). The level of absolute belief that being jabbed was a good thing because whatever symptoms anyone ever has, being jabbed protected them & caused illness to be less that otherwise would have been the case (I now have ‘headache’ from further banging head against the wall).
Dad did get a secondary infection, plus has ongoing water works problems and unfortunately got a non-covid water works infection which meant a trip to A&E and a course of antibiotics (which has previously sorted similar problems). Again, he is being coerced &, when in a vulnerable condition, the guard can drop.
If the worst does come to the worse with either or both, I shall have them on aspirin.
My experience of a mid-50’s with coronavirus – I’d have said it was influenza. Bit rough for 3-4 days (Tues-Fri), developing an annoying cough around the Thurs lasting around a week. Fully recovered.
The world is going mad – so sorry for the care home staff & NHS workers being affected by various mandates. Stay strong.
There does seem to be an assumption that those who have had a vaccine are better off than those without.
Whilst my example is only but an anecdote, this is what happened to us;
My wife, 53 years old, had two AstraZeneca jabs, I’m 65 and decided to have no vaccine. My son, 20 years old had 1 Pfizer jab and my daughter, 17 years old had no jabs.
Out of all of us, my wife seemed to suffer the worst symptoms. Sensitive skin, cough, headache, loss of taste and smell. Ill for around 9 days. My illness, was just like a very bad cold with a slight cough.
We also took, Vitamin D3, vitamin C, Vitamin K2 MK7, Zinc and Magnesium. We also took Andrographis Paniculata, this is being looked at as a possible treatment in Thailand and China.
All my wife’s work colleagues assumed not having a vaccine, and she would have been worse!
Perhaps she would, and perhaps she wouldn’t, I can’t prove it either way. I do though, think people could be more circumspect before making their proclamations.
As a family we have no co-morbidities, exercise and are reasonably fit and healthy.
Thanks for your anecdote Steve. I listen to people talk and those that are jabbed seem to get worse symptoms (though save for a small number of family members, I know of one unjabbed work colleague only) – even when someone jabbed get hospitalized it seems that the consensus is that the jab saved said person from an even worst outcome. Yes, it can’t be proved either way. When jabbing first started, there was plenty of discussion – it does not come into general conversation now.
I take similar (plus quercetin) & have family members doing same (I buy them and dish them around) – I’ll have a look into Andrographis Paniculata.
I now keep schtum about not being jabbed (I don’t want to ‘scare’ the jabbed). There is no requirement to get it with work and it is left to personal choice but new guidance has been published on the Intranet in October that it is strongly encouraged to get jabbed to attend the office (apparently to protect those that are jabbed (???)) & that line managers are to enquire about jab status. Needless to say I shall not be playing this game – I shall not reply to any emails and then if & when I am asked in person / Teams / phone, I shall stick to the ‘KISS’ principle (along the lines ‘decline and goodbye’).
As I’ve mentioned before, Thiamine Deficiency (Beri Beri) affects any aspect of ANS function, including heart function, breathing, digestion, etc & can trigger a whole raft of issues. Acute deficiency can radically affect the heart (wet Beri Beri) by whatever mechanism it uses, but many other symptoms can manifest before it gets to that point, such as anosmia & breathing difficulties (low or poorly utilised B12 can do that too. ‘Air Hunger’, inability to take full breaths & having to consciously breathe is one of my symptoms when my B12 is too low. I have to use ‘active’ sublingual B12 drops because oral tablet & capsule forms don’t work due to my gut damage & my body cannot convert it to the active forms for cellular uptake).
B vitamins including Thiamine are crucial for energy (ATP) production. When the draw on these nutrients is greater than the supply – such as during periods of stress, trauma & illness – then it is inevitable some functions will suffer. Low Thiamine & B vitamins in general is a big problem in our modern diet & stressful lifestyle. Contrary to being an ‘old’ disease, Beri-Beri is very much alive & kicking (our butts) in our modern ‘high-calorie malnutrition’ dietary life.
If you can obtain a copy, Dr. Derrick Lonsdale’s ‘Thiamine Deficiency, Dysautonomia & High Calorie Malnutrtion’ tome of reference is well worth reading.
With a second reading I am beginning to grasp the importance of RAAS in both covid and CVD, and this explanation why children are largely immune to both (limited ACE2 expression in children):
https://trialsitenews.com/covid-injections-offer-children-no-opportunity-for-the-benefit-and-only-potential/
This is also interesting (beta-blockers in covid treatment) in light of RAAS:
https://www.theepochtimes.com/heart-drug-shows-promise-for-covid-19_3997437.html
Vitamin D is some of the answer but not all of the answer. Compare the summer and winter waves in Gauteng province, South Africa. Located just outside the tropics at 26 deg south and an altitude of 1300 m, there is no shortage of summer sunshine, and shorts and slip-slops are the typical summer dress, so we can assume plenty of natural vitamin D in the population. It’s a crowded, cosmopolitan, go-getter and highly urbanised area with plenty of mixing, perfect conditions for Covid to spread.
Drat! Image of Gauteng waves didn’t display. Here is the link:
Sunscreen, advanced age, overconsumption of recreational drugs (especially ethanol), being a dark-complected office worker, and obesity defeat sun-induced vitamin D. Those are the groups I can think of off-hand–there are probably more.
Vitamin D supplementation is necessary for many for optimal serum 25OHD levels. Someone who is quite dark might require 12 times or more the sun exposure time of someone who is light-complected for optimal vitamin D production. Perhaps three hours in the sun. Then there’s the melanoma issue for dark-complected people which induces many of them to avoid the sun.
Perhaps doctors ought to be testing vitamin D levels once a year or more as a general rule.
Can I suggest you read Anderson and Grimes books on “Vitamin D Deficiency and Covid 19” at £6 it is a good relevant read, and it will answer some of your questions.
Fascinating!
Since spike proteins are present on some other viruses, perhaps the main difference between this Covid and others is its usage and importance in promoting and imposing a new world order.
Again, Caution.
‘Why does this conversion {from Angiotensin l to Angiotensin ll} occur in the lungs, not the kidneys or liver?’
Maybe because blood from the lungs has a direct route to the heart?
and a direct supply from the lung would be helpful because an increase in heart rate and blood pressure often needs to happen quickly?
“It is why, after I got vaccinated, I took aspirin for a month.”
Hey, what vaccine do you consider as the safest one to get and what doses of aspirin daily? I imagine small doze, similar to those in medicine for cardio prevention.
” I will even start to promote my new book – due to launch in October. ‘The enduring mystery of heart disease – The Clot Thickens.”
That’s a book to must-have then. 🙂
If you’re fit and healthy, active with no underlying health issues and under 80 years old, I’d suggest the best ‘vaccine’ is NONE. Of course, if your local branch of the Gestapo insists on jabs for your job, income, freedom then you will have to make an informed choice, but remember you cannot untake it.
“if your local branch of the Gestapo insists on jabs for your job, income, freedom then “ then you should remind them they will be complicit in a crime for which they will be personally liable, and there will be no legal protection for them. “Nuremberg” may wake them up.
They won’t care. They don’t believe they’ll be prosecuted. They are probably correct.
Video all about RAAS (Renin-Angiotensin Aldosterone System) – It’s possible link to SARS-CoV-2 and mRNA vaccine. An education, she explains it very well and suggests other researcher get involved!
Dr. Jessica Rose (PhD, MSc, BSc)
Take home message – The spike proteins produced by host cells may interfere with RAAS
SARS (SARS-CoV-2) why is it bad for some people?
If you already have an overactive RAAS system (eg: the elderly) SARS-CoV-2 has the potential to throw a wrench and make it worse
1. Renin-Angiotensis System
2. SARS-CoV-2 involvement
3. mRNA spike protein involvement
Conclusions
The RAAS is compromised to induce serious pathologies by SARS, especially in individuals with existing over-active RAAS
Similar pathologies may arise due to the presence of exogenous spike proteins ie mRNA Pfizer vaccine
Inadequate ACE function is an independent predictor of mortality
Aguesia and anosmia (loss of taste) have been associated with RAAS
Adverse Events associated (cardiac, bleeding, inflammation, oxidative stress, kidney disease) with RAAS dysfunction are frequently being reported in the context of COVID-19 injections
An additional report by Dr Jessica Rose
VAERS report for VCC
If people on this site have not done so before, may I suggest that they hop over to John Dee’s Almanac on FB as he’s starting to release his analyses of nhs data. The first tranche makes devastating reading.
I will not be the only one who can’t do that. I have nothing to do with Facebook .
You can read all his work by signing up to Pocketnet. This is the backup channel when FB decides John Dee is getting too close to revealing the truth.
Clarification sought re ” coagulation and immune system”, “and inflammation and hemostasis (blood clotting)” Is there a difference between coagulation – in this context, and hemastosis ?
Thanks in advance
Excerpt about scientific studies from the US Senate testimony by Harvey Risch:
“These studies break down into two major types. The first is double-blinded, randomized
controlled trials, and the second is non-randomized but still controlled trials. You have heard
from various government and scientific personalities that randomized controlled trials provide
the strongest form of evidence. Many of these people have also claimed that randomized trials provide the only trustworthy form of evidence. There is some truth in these assertions, but
there is also lots of falsehood. We know for example that the great majority of drugs used to
treat heart diseases were established with non-randomized trials. Cholesterol-lowering drugs
were in widespread use before randomized trials were ever done. Azithromycin, the most
commonly used antibiotic in children, was not established by randomized trials. The idea that
only randomized trials provide trustworthy evidence is a simplistic notion that may sound good
in theory, but the comparison between randomized and non-randomized trials is something
that has actually been extensively studied in the medical literature. I am an epidemiologist
because even though I love biological theories, I develop them all the time to study how nature
works, but it is from the human empirical data that we learn how indeed nature works.”
Risch goes on to reference the Cochrane reviews comparing retrospective and prospective studies and finding their reliability to be comparable.
Additionally, you might want to read this short article on Dr David Healy’s blog (UK psychiatrist) on the history of RCTs and why they are a gift to the pharmaceutical industry https://davidhealy.org/clinical-trials-are-unsafe/ Also look at the links
John: Thanks you for that link! This is something everyone should read.
Oh, yeah, RCT foundationalism is one of the main mechanisms by which pharma captured medical research.
Could a reader please explain why Trudeau, Morrison, Biden are demonising the unvaccinated, saying now it is the ‘pandemic of the unvaccinated;’ also, in Morrison’s case saying if one gets CV19, you will die!! Is there a ‘plant’ from the WHO/Gates Foundation giving instructions? We have truly lost our sovereignty.
If one can read other languages you will see that there are exact similarities happening there too. My brother still thinks its incompetence by the UK government when I can see its more a global tyranny all aping the same words.
Dana, not the only reason, but one reason is that those who are jabbed and/or double jabbed who die within a few weeks of arm spearing, are classed as “unvaccinated”. The morons you mentioned are also under control of the elites, and will say anything they are told to as they have no useful knowledge of their own.
I saw a video by a Belgian psychologist about this phenomenon. Basically, the unvaccinated are in the role of the target group and Faucist leaders are demonizing them–similar to the roles of the Jews and Nazis in WW2.
I believe it is a game of political one-upmanship. “My people are more vaccinated than your people, therefore I am a better politician than you. Nyaaah nyaaah nyaaah.”
It’s a great pity that ‘percentage vaccinated’ is the number that people use to keep score. It has nothing to do with the overall health of the population. It is rather like a cardiologist bragging, “I have prescribed more statins than you, therefore I am a better cardiologist.” when we know it is how many heart attacks has he prevented at what cost, which involves stress, nutrition, exercise, impact on family etc, not just the amount of medication.
A better measure might be = increase in unemployment %+ excess deaths% + reduction in GDP growth rate % + businesses closed%. But of course that is rather difficult to manage.
What really concerns me is the willingness to keep imposing restrictions and lockdowns until a certain percentage vaccination is reached. This is penalising the many for the ‘sins’ of the few vaccine-hesitant. It’s collective punishment, which is illegal.
The punished group may often have no direct association with the other individuals or groups, or direct control over their actions. In times of war and armed conflict, collective punishment has resulted in atrocities, and is a violation of the laws of war and the Geneva Conventions. — https://en.wikipedia.org/wiki/Collective_punishment
Meant to say …rather difficult to measure.
Some websites say not to use Aspirin for Covid- as the type of clots are different (Aspurin widening vessels) whereas Covid coagulates the blood. So I was suprised to hear you took Aspirin. Can you please discuss and perhaps in what quantity.
To improve your odds if vaxxed, take nutritional supplements…
” Computational systematics of nutritional support of vaccination against viral and bacterial pathogens as prolegomena to vaccinations against COVID-19″
Could you please explain a bit more about aspirin. I have been told the blood clots associated with Covid and their vaccines (coagulation of blood) are different from normal blood clots (constriction of blood vessels). Why take aspirin then?
You have been told?
Reports in the media suggest not to take aspirin. Its a different type of clot. Can you please explain in your blog why aspirin works as a preventative of strokes if you have Covid or are vaccinated
Dear Dr Malcolm,
I have a question which I can see you partially responded to on another comment, a suggested dose of 75mg aspirin. I also wondered if you were able to share the frequency you took the aspirin? One tablet daily?
I also wondered if aspirin could be suggested in the same way to prevent/reduce risk of clotting when infected with covid? Eg taking aspirin for month after infection. If you have any thoughts I’d be very grateful. Thank you for your blog.
I had a heart attack. I am a 55 year old thin woman with only one risk factor, high ldl. I exercised daily. I had the heart attack in January at the height of a covid surge. I was negative for covid. I think I had covid in 2019 before there was a test. I was sick for 3 months with what I was told was a sinus infection. When I had the heart attack, the doctors found an old blood clot in my leg. I now have 4 stents and I am on cardiac drugs.
If you read Tom Levy’s work, it may well be the sinus infection had a significant bearing. There is information that infections above the shoulders have a lot to do with heart problems.
RTE ‘ the irish state broadcaster which has been a stalwart defender of government mandates and of the pharma conglomerates’ premature cure has changed its spots – so to speak @joeliveline wants to hear from anyone that has suffered from adverse reactions from the COVID-19 jabs.
Wow, if toes end up looking like that, I don’t want to know what blood vessels look like.
https://www.theguardian.com/world/2021/oct/06/covid-toe-may-be-side-effect-immune-response-study
Nobody in this household or anyone else I know who tested positive hav been so affected. Are we just the lucky ones or os this a headline that has found an ailment – a topical ailment that is. Sort of nay sayer’s readers digest article. Anybody remember the RD ?
Is that the point we got to? So do we only believe in things we have seen ourselves?
No. But we begin with observations. Is this experience common or an aberration? What is the frequency of “covid toe” ?
It’s the first I’ve heard of it and the article does hav a tabloid feel to it.
I understand, nothing that a little ‘google’ couldn’t solve: https://www.aad.org/public/diseases/coronavirus/covid-toes, https://health.clevelandclinic.org/are-covid-toes-and-rashes-common-symptoms-of-coronavirus/, https://jamanetwork.com/journals/jamadermatology/fullarticle/2767775. Not a common symptom, but also not rare. It seems that it’s not uncommon for viruses to cause rashes. Does that help you?
Did that answer your first question – is this a new thing? The answer is no (First publication from April 2020)
As for your second question – Do you know anything about this? I guess also no
Thanks David. My armchair research suggests any corona associated virus can result in covid toe.
“Dermatologists warn that “COVID toes” may be a first or only symptom of a coronavirus infection”
https://www.health.com/condition/infectious-diseases/coronavirus/covid-toes
Also in my travails i have come across “covid tongue” – courtesy of the indiana express.
But might covid toes be as a result of exposure to severe cold ?
https://www.aarp.org/health/conditions-treatments/info-2020/unusual-coronavirus-symptoms.html
Went searching further, wondering to myself, could there be covid haemorrhoids?
Look what I found.
https://pubmed.ncbi.nlm.nih.gov/33113796/
Yes. Dreaded covid has a haemorrhoidal ring to it too. No end to its ferocity.
And just as I was about to end my research, I discovered that the vax too may induce haemorrhoidal activity.
Definition of disease
noun: disease;
a disorder of structure or function in a human, animal, or plant, especially one that produces specific symptoms or that affects a specific location and is not simply a direct result of physical injury.
Hmm, specific symptoms …
Looks like the definition of disease needs changing now because of the ,myriad symptoms attributed to “covid” …
MG
Sorry for popping up again, but you answered my 2nd question with a question.
One of the earlier symptoms.
No, a lot of commenters believe the bs of Alex Jones, Donald Trump etc.
Alex jones ? ??
And when is last time u heard anything from Trump. Still, made lots more sense, as bad and all as he was, that the new chap.
I used to say that too – I hadn’t seen anyone dropping dead Wuhan style in the street until very recently …
It’s a common occurrence round these parts now too … falling down like flies they are …
Now where did that influenza mortality go … must have put it somewhere …
MG
Next up – all you ever wanted to know about covid haemorrhoids but were afraid to ask.
Looks like the Russkies are at the forefront again!
Read this post from Dr Kendrick yesterday and saw this today..
https://www.rt.com/russia/537205-new-covid-drug-cytokine-storm/
“This is an evolutionary mechanism we inherited from our ancestors,” Skulachev said, adding that the evolutionary trait hasn’t caught up with the fact that humanity has developed vaccines and medicines to deal with such diseases.
“Leitragin is aimed at suppressing inflammation processes,” Karkischenko said, adding that “it is a drug based on an absolutely innovative approach” and can be used to treat “various conditions.” The FMBA center is already working on different drug formulations for Leitragin.
This is yet another example of the human race in thinking they can do better than nature. Handy when fixing broken bones, which is simple, but not when it comes to the complexity of the immune system.
Hi Jdafae: rats have not evolved to cope with hyperglyemia
https://journals.lww.com/ccmjournal/Abstract/2008/08000/Effects_of_hyperglycemia_and_insulin_therapy_on.28.aspx
Effects of hyperglycemia and insulin therapy on high mobility group box 1 in endotoxin-induced acute lung injury in a rat model
“Abstract
Objective:
Hyperglycemia and insulin resistance are commonly seen in septic patients and are associated with increased morbidity and mortality. High mobility group box 1 (HMGB1) protein has been shown to play a key role as a significant factor in sepsis pathogenesis. This study investigated the increase in lung damage because of hyperglycemia and HMGB1 increase in a lipopolysaccharide-induced septic rat model and the potential for insulin therapy to reduce this lung damage by decreasing the serum level of HMGB1.”
“Conclusions:
Hyperglycemia is associated with higher HMGB1 levels and lung damage in sepsis. Insulin therapy significantly reduced lung damage, suggesting that management of hyperglycemia with insulin might decrease HMGB1 levels in the serum and lung tissue. One of the mechanisms that could contribute to the inhibition of HMGB1 secretion might be related to the inhibition of NF-κB.”
You got vaccinated 🤔
Sent from my iPhone
Kinell Anthony, I wouldn’t normally reply to these but the emoji thingybob that you use and the ‘I’ve got an iPhone’ (should I let you mine?) {further apology as one of my asides – it is Saturday night I’m enjoying some of the 40% produced and bottled in Scotland stuff though I’d argue it is 60% water [I do further apologise to the Whisky aficionado’s that this is the blended stuff and my excuse is that it is on offer in England with a particular storecard}.
Anthony, if you have been following Dr K’s blog, Dr K has made it clear right from the get-go his reasons for being vaccinated from coronavirus and which are very noble. I’m sure that you could have done searches of the blog to find the reasons before posting your question (& which could have avoided the question with the silly emoji) – I won’t spoil your fun searching but for Dr K, doing an NHS job and dealing with vulnerable patients in care homes, it is a selfless & unquestionable act.
I do not care whether someone is / is not vaccinated against anything. What I do care about is people questioning whether someone is / is not vaccinated against whatever – it is their business (I’ll stop here having deleted what I was going to say).
Personally I am not jabbed against coronavirus (my risk / benefit analysis is that risks outweigh the benefits – though I am open to change my mind if [brackets, big IF] future data to the contrary becomes available). I am happy to provide further details, if anyone is interested {but have deleted as I don’t’ want to bore anyone further} –might be the 40% vol. & Scotland’s fault.
P.S. COP26 in Glasgow ends on Friday 12th November 2021 – if you need to get anything sorted out, might not be a bad idea (for the UK based) to do this by COP26 end date (influenza might start kicking in after COP26).
The quote about spike proteins and platelets does not currently appear in the article linked by footnote 7.
I found it. The quote attributed to the article in footnote number 7 is actually from this article:
“The roles of platelets in COVID-19-associated coagulopathy and vaccine-induced immune thrombotic thrombocytopenia”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8390120/
Anyone care to comment on the possibility of the pfizer vaccine containing graphene oxide, and how to detox this substance? I have read kaolin clay will detox this. Apparently the graphene oxide can cause a multitude of disorders.
I’ve seen a few articles which suggest there may be vaccine’s containing graphene oxide, but I have serious doubts.
What has grabbed my attention are the lipid nanoparticles in the Pfizer vaccine? They use PEG (polyethylene glycol) to deliver the product. PEG does pass the blood brain barrier
One of the problems of delivering drugs to the brain is the blood brain barrier, so PEG is being considered. This article explains it quite well – https://www.materialstoday.com/biomaterials/news/polymer-nanoparticles-on-the-brain-/
AstraZenneca uses Polysorbate 80 which can also pass the blood brain barrier.
The drug companies claim there are insufficient quantities of these lipids to cause any problems. Drug companies are of course perfect (not) so we should trust everything they say without doing our own research!
Well I read about this here: https://www.redvoicemedia.com/2021/07/deadly-shots-former-pfizer-employee-confirms-poison-in-covid-vaccine/
The variety of adverse affects is so great one might think there is more than just the spike protein at work…PEG, Graphene oxide…what else…
Does Dr. Kendrick have any comments/ opinion on the new COVID 19 therapeutic pill?
Is there any truth in the new Covid 19 soothing linement balm, from,Lemsip? Mandatory for all and accessed at various church halls, car-parks and primary schools located around the UK?
So the message is let’s bow down, be broken in our soul. Let’s accept tyrannical impositions upon us whenever the authorities please and in the name of any potentially made up excuse. But, let’s take some aspirine to try to amelliorate the unknown effects in our bodies long term.
What can we take for our broken wills and spirits, and our painfully gullible and weak minds good doctor?
Buy a copy of “The Clot Thickens” and learn how to fight back. Truth is knowledge, and knowledge is power. The important bits start on page 5.
The reference #7 to the quote starting with “Thrombosis is a major pathological driver in COVID-19…” doesn’t seem to contain that paragraph. But I’d like to use that referenced paragraph. Can that be checked ?
Found the quote ref #7 is actually from
https://www.sciencedirect.com/science/article/pii/S1050173821000967
Doctor,
What is your take on the abstract published by Gundry (https://www.ahajournals.org/doi/10.1161/circ.144.suppl_1.10712)?
The publication has now published an ‘Expression of Concern’, citing a number of error: ‘Specifically, there are several typographical errors, there is no data in the abstract regarding myocardial T-cell infiltration, there are no statistical analyses for significance provided, and the author is not clear that only anecdotal data was used.’
Dear Dr.Kendrick,
I came across this article recently. I was wondering, whether you would like to comment on it.
I have some hard nose anti vaxers in my family, once or a while I still try to convince them to get their shots!
Now with this article, they and their friends in the same bubble are convinced that mRNA vaccination damages your DNAâ¦
BTW, I just finished your book âThe clot thickensâ â very interesting, though Iâm not in the medical profession. I need to read it again.
Best regards
Dr. J. Harter
Von: Dr. Malcolm Kendrick Gesendet: Donnerstag, 16. September 2021 10:25 An: jfharter@aol.com Betreff: [New post] COVID19 and CVD â Bridging the gap
Dr. Malcolm Kendrick posted: ” 16th September 2021 Bridging the gap between cardiovascular disease and COVID19 [Where two diseases meet] Having announced that I will not discuss COVID19 anymore, I am about to do so â at least in part. Yes, you may now be thinking⦠how can we “
and where is you position on injecting children doc?
Sounds like the cue for a song (a suitable tune will come to mind)
Jab ‘em all,
Jab ‘em all,
The long and the short and the tall,
Jab all the oldies, the dads and the mums,
Jab all baby daughters and all baby sons,
This is the sick world of today, with far too many people believing in this false god religion. It must be total rubbish as it is pushed as a one size fits all fix, for a relatively trivial problem.
Jfharter, yes, get all the people you can to get their shots, but first get them to read, and take note of the encouraging words from Stefan Oelrich https://pharmaceuticalfraud.com/2021-11-18-covid-mrna-shots-gene-therapy-falsely-marketed-vaccines.html. That way you cannot be accused of bias. Are you a medical doctor, a GP perhaps?