THE WAR
When I first heard the CTT at Oxford had published their latest analysis on statin adverse effects I must admit I sighed deeply. Do they never give up? This was the latest salvo in a long running war between the pro-statinators and the anti-statinators. Or, as I like to think of it, those who are wrong, and those who are right.
Why is there such a war? Because, and for reasons that I still find difficult to fully comprehend, statins have reached the position where they are believed to be the miracle cardiovascular drugs. With uniquely powerful lifesaving properties. ‘All hail the mighty statin’.
A friend was recently told to take atorvastatin. She was additionally told that if she did not, she would most likely die of a stroke or heart attack. Does anyone believe this is a reasonable thing for a doctor to tell a patient? So much for a therapeutic, non-paternalistic, healing partnership.
I cannot think of any other medical intervention where you get the added threat of oblivion thrown in for good measure. I suppose it happens with blood pressure lowering tablets from time to time. Maybe a few others. Chemotherapy? Vaccines, certainly, but that is a whole different ball game.
With most other drugs, if you think they are causing problems, the doctor will be reasonably happy to switch, or even stop. With statins there is a relentless pressure to keep on taking them. No matter what.
It is true that you may be allowed to move around between various statins, but as for stopping completely. You will be most likely be told this, or some version of it. ‘If you stop taking them, you will have a stroke or heart attack, and die!’
I have read countless e-mails from patients who have been told by their doctor that they will be thrown of the list if they refuse statins. Not from the UK, but in the US. Where it seems easier to fling people off your medical list. How nice it must be to have such, responsibility free, power. ‘I shall deign to treat you, so long as you do exactly as I command. Look upon my works, ye mighty, and despair.’
On a wider scale, if you talk to the media, or even write peer-reviewed articles suggesting that statins are anything other than miraculous life savers, you will be relentlessly attacked. I speak from personal experience, and a five year long libel trial.
Others have lost their jobs. Uffe Ravnskov, a long-time statin critic had his book The Cholesterol Myth burned, live, on air, in a TV studio. Dr Maryanne Demasi was booted off her job on the Australian Broadcasting Company. Then, for good measure, they tried to strip her of her PhD. Pour encourager les autres, I presume.
Kilmer McCully was flung out of Harvard for, in essence, pursuing ‘non-cholesterol related’ ideas on heart disease. His treatment was commented on by Thomas N James in a New York Times article. Kilmer was attacked in the 1970s, so this goes way back.
Thomas N. James, a cardiologist and president of the University of Texas Medical Branch who was also the president of the American Heart Association in 1979 and ’80, is even harsher [regarding the treatment of McCully]. ”It was worse than that you couldn’t get ideas funded that went in other directions than cholesterol,” he says. ”You were intentionally discouraged from pursuing alternative questions. I’ve never dealt with a subject in my life that elicited such an immediate hostile response.” 1
Just what the hell is it with statins and cholesterol lowering? I think I know this area as well as anyone, if not better than most. But I still find it difficult to work out where the level of conflict and aggression comes from. The polarisation of views.
It seems to go well beyond all rational behaviour. When two tribes go to war? Or maybe, to quote Cass Sunstein, author of Nudge. ‘To become an extremist, hang around with people you agree with.’
The benefits of statins – as I see them
I have studied the benefits of statins for many, many years. Probably too many, in truth. And, assuming the clinical trials were not entirely fabricated, I agree that they have some benefits. I do not think they are great, but they exist.
Can they be fully quantified? I could end up quoting paper after paper at this point, but I will restrict myself to two, and a bit. The first is a meta-analysis of twenty-one statin trials published in the Journal of the American Medical Association. It was pretty thorough, and one of the most recent – that I am aware of.
They found that statins led to a reduction of:
- 0.4% for stroke, fatal and non-fatal.
- 1.3% for myocardial infarction – fatal and non-fatal
- 0.8% reduction of dying (of anything)
These benefits accrued after about four years of taking a statin, on average. Sticking to overall mortality at this point, and using their 0.8% reduction figure, you could expect to see a reduced risk of death of around 0.2% a year. 2
Which is clearly important, but only in so far as it goes. I say this because the 0.2% figure certainly does not represent the end of the story. It is not enough to know how many more people were alive when the study ended. It is, in my opinion, far more important to establish what this means for an increase in life expectancy.
Reducing risk of death, and increasing life expectancy. No, they do not mean the same thing – at all. Even though they sound as though they probably should. And, while there is clearly a relationship between the two – it may not be what you think.
To use an extreme example. CPR may stop you dying of a heart attack on Tuesday, only for you to suffer a second heart attack, then die, on Wednesday. Or it could stop you dying on Tuesday, and then you live happily for another forty years. It is important to know which of these is most likely to happen – on average. One day, or forty years.
My view is that the effect on life expectancy is the single most important outcome we have. Especially when you are looking at an intervention designed to ‘prevent’ future events from occurring.
In cancer this is what used to be measured, and presented. The main reported outcome was, ‘increase in median life expectancy’. Which is not quite the same thing as average life expectancy, but … for the sake of this argument, it is close enough.
If a cancer drug is found to increase life expectancy by, say, around six months in a clinical trial, the patient will be told, something on the lines of…’if you take this drug it should give you another six months of life.’ [Cancer trials have now moved away from this endpoint, instead they look at other things, such as progression free survival (PFS). This has not been universally welcomed, other than by the pharmaceutical industry*]
Increase in life expectancy has never has been an outcome for cardiovascular trials. Why not? Well, there are valid statistical reasons. Median survival means the time it takes for fifty per cent of the trial population to die, in both arms. Drug vs. placebo, or new drug vs. standard management.
Sadly, this can happen rather quickly in cancer trials. But it could take twenty or thirty years to get to this point in a cardiovascular trial. And no-one is waiting that long. Apart from anything else, the patent protection for the drug would have run out before the trial finished.
Instead, in a cardiovascular trial, they (may) tell you the percentage reduction in death at the end of the trial – if there was one, which there often isn’t. But how does a reduction in death relate to life expectancy? Can a figure be established? Well it can, if you take a few more steps.
Below is a simplified graph from the Heart Protection Study (HPs), looking at the survival curves for statin or placebo. At the start everyone is alive, as you would expect. By year five, more people have died in the placebo arm than the statin arm. I think I should mention that this was one of the most positive statin trials ever done. This is me, giving them the benefit of the doubt.
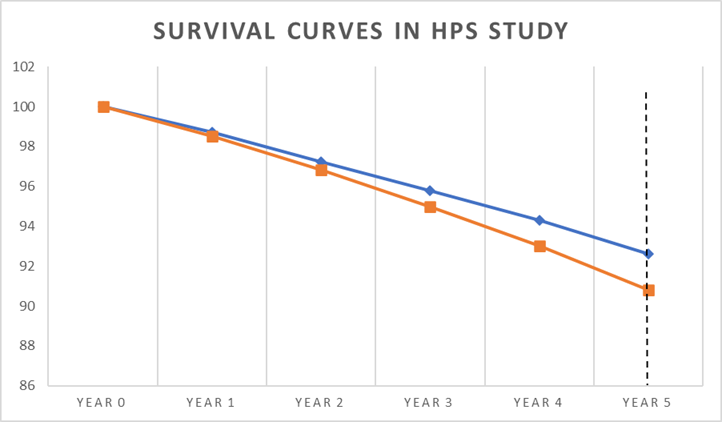
The vertical dotted line at year five represents the end point of the study, where you can see there is a clear difference in the percentage of deaths:
- 90.8% were alive on placebo
- 92.6% were alive on statin (simvastatin)
A difference of 1.8%.
The press release for this trial stated the following:
“In this trial, 10 thousand people were on a statin. If now, an extra 10 million high-risk people worldwide go onto statin treatment, this would save about 50,000 lives each year – that’s a thousand a week.”
Rule number one of medical research, you cannot save lives. Unless you succeed in making people immortal. The most you can possibly achieve is to increase life expectancy…by some amount.
Indeed, ‘life saving’ is a completely non-scientific term that should not be used to describe any clinical study. Although, obviously, it does sound rather good. Fifty thousand lives saved a year has a certain ring to it which, you could argue, 0.2% does not.
Moving on. In the HPS study 1.8% more people were alive after five years. But how much longer, on average, would they live. One day, or forty years? Well, you cannot calculate this precisely, due to the somewhat wobbly nature of survival curves, but you can get close enough.
The first step in doing this, and the most simple, is to draw a horizontal line through the survival curves, rather than using a vertical one at the end. This is effectively what they do, or did, with cancer trials. Although it is a little more scientific than that.
In the diagram below, I have drawn my horizontal line at four years, taking it from the placebo survival curve. If I drew it at five years, I would be drawing a line out into empty space.
After four years, 94.3% were alive in the statin arm, and 93% on placebo. A difference of 1.3%. How long did it take for those in the statin arm to reach the 93% survival point seen in the placebo arm? Well, as you can see from where the arrows are pointing it was less than a year. About nine months, give or take.
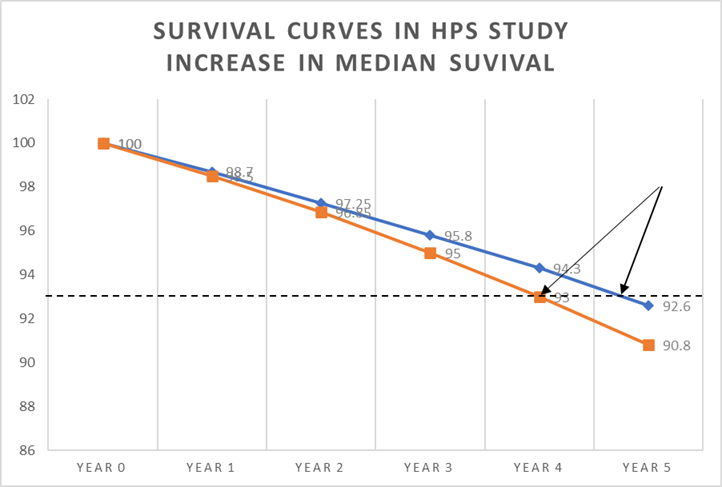
Moving on to the next step, we can now say that, on average, the extra 1.3% who were alive after four years would then live an extra nine months. Which allows us to make the following calculation.
Average increase in life expectancy after four years is nine months (274 days)
But this is not for the entire population, it is only for 1.3% of population.
So, to establish the average life extension for the entire population we need to multiply 274 days by 1.3%
274 x 1.3% = 3.5 days [274 x 0.013 = 3.5].
After five years, it is likely this figure would increase by five fourths. Divide by four and multiply by five.
3.5 days 4 x 5 = 4.4 days
Clearly these figures are not precise, but they are close. If you look at the HPS study in this way it means that if you take a statin for five years, you may expect to live for an extra 4.4 days.
There are other ways to do this calculation, but they all come to pretty much the same conclusion. I once did the calculation using a much more accurate method called Reduction in Mean Survival Time (RMST) where you calculate the area under the curve for the statin and placebo, then look at the difference, then do a bit of mathematics. But I could not get it published anywhere.
One study in the BMJ Open, and the only one to do this, as far as I know, did look at the life expectancy issue, taking data from a number of statin trials. They worked out the increase in life expectancy – if you take a statin for five years – which is how long many of the major statin trials lasted.
The researchers then split the results in two.
- In primary prevention the average life extension was 3.2 days
- In secondary prevention the average life extension was 4.1 days.
Which, as you can see, is pretty close to my simplified calculation. [The HPS study was a secondary prevention trial]. 3
This figure certainly caught the attention of the CTT at Oxford. Their response can be summed up by Professor Colin Baigent in the Mail on Sunday article – the article that I sued them for.
‘…the important thing is statins extend healthy life. They avoid both disabling events like heart attacks and strokes. Simply focusing attention on extra duration of life is to ignore the fact that these drugs reduce disability.’
At first sight this is a valid point. But there are a couple of major issues to consider here. The first is that a ‘non-fatal’ heart attack is not necessarily disabling, in the least. They can pass virtually unnoticed. As do many strokes.
Despite this, in the clinical reporting on statins trials, no distinction is made between a crippling stroke, and one that leaves no trace it ever took place. Same for heart attacks. A stroke is a stroke, a heart attack is a heart attack What about: mild, moderate, severe? Nope, they all get bundled up together under the same heading. Which is clearly, and most probably deliberately, misleading.
However, and more importantly, the Baigent argument is effectively negated by the data I presented in the previous blog. Which is that, while ‘serious cardiovascular events’, such as a stroke, are reduced, other Serious Adverse Events (SAEs) are increased – to the same degree. These can be equally damaging and dangerous.
If cardiovascular serious adverse events are viewed in isolation, 71 primary prevention patients with cardiovascular risk factors have to be treated with a statin for 3 to 5 years to prevent one myocardial infarction or stroke.
This cardiovascular benefit is not reflected in 2 measures of overall health impact, total mortality and total serious adverse events. Therefore, statins have not been shown to provide an overall health benefit in primary prevention trials.’
Their conclusion: unrecognized serious events are increased by statin therapy
This, then, is the lens through which I view the benefits of statins. They do not reduce ‘overall’ serious adverse events, and they increase life expectancy by around eighteen hours… a year.
Adverse effects vs efficacy – the interconnected battle
If you view statins through the lens that I do, then the rate of adverse effects is almost completely irrelevant. Why would anyone put up with any unpleasant effects to achieve such a limited benefit?
In addition, you could easily spend your extra eighteen hours a year getting your cholesterol level checked, picking up the prescription, taking the tablets, waiting to get the prescription filled at the chemist, visiting your doctor. Worrying about your cholesterol level.
Clearly, however, my position is not widely shared. Here are a couple of quotes from the researchers who did the CTT Oxford study on adverse effects.
‘Oxford University researchers said decades of misleading information may have put millions off taking the daily pills. They can slash the risk of heart attacks and strokes by up to half for those at the highest risk.
50% of patients who would benefit from these extraordinary drugs are not receiving them, for whatever reason.’ 4
These ‘extraordinary’ drugs.
Yes, here is the heart of the interconnected battle. Benefits vs. adverse effects. In truth, you can’t really talk about one without paying close attention to the other. If the benefits are great, people will be willing to put up with many more adverse effects, and vice-versa.
Before having read this article you may have wondered how an Oxford researcher can state ‘They (statins) can slash the risk of heart attacks and strokes by up to half.’ Yet, I am on record as saying that statins are really not that great. When you look closely, they increase life expectancy by eighteen hours a year, and that’s about it.
Do we live in a weird parallel Universe? Am I lying; are they lying? Nope, as I hope to have demonstrated, we live in the same universe, we simply choose to frame the data in a different way.
A few years ago I wrote a book called Doctoring Data – which looks at the games played with medical research. I quoted Professor Michael Baum on prostate screening.
‘Every year I play a game with the senior postgraduate students at a course for specialists in cancer run by the Royal College of Surgeons in England. I tell them that there are two potentially effective screening tools for prostate cancer. One of which will reduce their chances of dying from the disease by between 10 and 30 per cent, while the other will save one life after 10,000 years of screening.
As a consumer or as a public health official, which one would you buy into?
They all vote for the first; yet the programmes are the same, they were just packaged differently.’
So, yes, it is scientifically correct to claim a 10 to 30 per cent reduction in prostate cancer deaths from screening. This is not a lie. It is a statistical fact. However, precisely the same data tells us that you can save one life after 10,000 years of screening. Or, to be a pure pedant, delay one death. This is not a lie either, it is a statistical fact.
The pro-statinators claim that statins slash the risk of heart attacks and strokes by up to a half (in truth, this is really pushing it. One study, stopped short, somewhat dodgy gathering of data, but I shall let it pass). I say, take a statin for a year and live eighteen hours longer. We are both right. Or correct, or whatever word fits best here.
It is up to you, I suppose, to decide who you want to listen to. What you wish to believe. What you think is most important.
Perhaps all clinical trials should be mandated to report in the same way, using all possible forms of outcome measurement. Here is a selection that you could include from cardiovascular trials
- Relative reduction in primary outcome e.g. fatal heart attacks
- Absolute reduction in primary outcome e.g. fatal heart attacks
- Relative reduction in overall cardiovascular events, fatal and non-fatal
- Absolute reduction in overall cardiovascular events, fatal and non-fatal
- Reduction in overall mortality, relative and absolute
- Number needed to treat (NNT)
- Number needed to harm (NNH)
- Reduction in composite end points – many different outcomes added together
- Serious adverse events
- Serious cardiovascular adverse events
- Serious non-cardiovascular adverse events
- Drug related events
- Average increase in life expectancy etc. etc. and on and on ….
At present there is no standardised way to report data from a clinical trial. You can present it pretty much any way you want. And, to a great extent, also create any message you want. To quote the paper. ‘How statistical deception created the appearance that statins are safe and effective in primary and secondary prevention of cardiovascular disease.’ 5
- The almost exclusive presentation of data in the relative risk format by statin advocates has intentionally misled the public to exaggerate the miniscule benefits of statins.
- Primary-preventive cholesterol-lowering trials have not succeeded in reducing the rate of mortality.
- The absolute risk reduction of CVD mortality in secondary-preventive cholesterol-lowering trials is quite small, rarely exceeding two percentage points, and no primary-preventive trial has ever succeeded in prolonging the life of the participants.
- The rate of serious adverse effects of statin treatment is highly underestimated.
- Adverse effects of statins are extensive, including diabetes, cognitive impairments, cancer, cataracts and musculoskeletal disorders.
- The small benefit seen in the cholesterol-lowering trials is independent of the degree of cholesterol lowering
Next, and finally on this damned topic. Why the CTT Oxford paper was so horribly misleading. A true masterclass in statistical obfuscation.
* Oncologists and cancer patients generally agree that the primary goals of advanced cancer treatment are to lengthen and/or improve patient survival. Yet over the last two decades, clinical trials of new cancer treatments have moved away from measuring outcomes that matter to patients… Research has shown that when patients are fully informed about the meaning of PFS (progression free survival), about half would not choose additional treatment for any magnitude of gain in PFS in the absence of an overall survival improvement’ 6
1: https://www.nytimes.com/1997/08/10/magazine/the-fall-and-rise-of-kilmer-mccully.html
2: https://jamanetwork.com/journals/jamainternalmedicine/fullarticle/2790055
3: https://pubmed.ncbi.nlm.nih.gov/26408281/
4: https://www.express.co.uk/news/uk/2166863/statins-side-effects-new-report