
16th September 2021
Bridging the gap between cardiovascular disease and COVID19
[Where two diseases meet]
Having announced that I will not discuss COVID19 anymore, I am about to do so – at least in part. Yes, you may now be thinking… how can we believe anything this man says?
However, I do have an excuse for this. Because, as part of my transition back to more familiar waters, I am going to look at the links that COVID19 has to cardiovascular disease… my life-long obsession.
The reason is that I have found it amazing how two apparently unrelated diseases can be linked so closely, and greatly increase your knowledge of both.
I will start with a quote that I would like to you read slowly, and carefully, taking a little time to think about – if you can get through the jargon.
‘Host defense against infection is based on two crucial mechanisms: the inflammatory response and the activation of coagulation. Platelets are involved in both hemostasis (blood clotting) and immune response. These mechanisms work together in a complex and synchronous manner making the contribution of platelets of major importance in sepsis. This is a summary of the pathophysiology of sepsis-induced thrombocytopenia*, microvascular consequences, platelet-endothelial cells and platelet–pathogens interactions.’ 1
*thrombocytopenia = drastic fall in platelet levels (small cells that conduct the entire blood clotting orchestra).
Yes, as you may have noticed, this passage says nothing about COVID19. On the face of it, it has nothing to do with cardiovascular disease either. It also contains a lot of jargon which most people without a medical background will struggle to understand. To me, however, it is fascinating, as it opens an entirely new way of thinking about critical disease processes.
What these researchers are saying, in the typically impenetrable prose of medical writing, is that the immune system, and the blood clotting (coagulation) system, have been designed to work together to fight off infective agents. Indeed, from an evolutionary perspective, they started off as the same thing. As discussed in an article in the Journal ‘Immunity’. ‘The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1α by Thrombin.’
‘Ancient organisms have a combined coagulation and immune system, and although links between inflammation and hemostasis (blood clotting) exist in mammals, they are indirect and slower to act. Here we investigated direct links between mammalian immune and coagulation system….The identification of a direct link between the coagulation system and the activation of the IL-1α* inflammatory cascade raises important questions.’ 2
*Interleukin 1 alpha (IL-1α) also known as hematopoietin 1 is a cytokine** of the interleukin 1 family that in humans is encoded by the IL1A gene. In general, Interleukin 1 is responsible for the production of inflammation, as well as the promotion of fever and sepsis. [Which is why you get hot and shivery when you get infected]
**a cytokine is a small protein that normally passes messages from cells to other cells and the immune system. Cytokines are key players in the immune response to infections, and there are many of them.
Anyway, put at its simplest. If you become infected (with almost any micro-organism,) you are far more likely to produce blood clots. Why? Well, it is probably because serious and life-threatening infections will often enter the body through a wound, or damage of some sort. Therefore, it makes sense that the body tries to seal off such wounds, or entry points, with a blood clot. This will not only stop the bleeding, but it will also trap the invading bacteria and viruses to prevent them spreading.
At which point the immune system gets to work on the trapped micro-organisms. Indeed, what better way to neutralize a virus, or bacteria, than by wrapping it up inside platelet fibrin complexes – two of the main constituents of blood clots?
At this point you may well ask, so what has this to do with cardiovascular disease, atherosclerosis and atherosclerotic plaques? Well, as the same paper goes on to say:
‘Many diseases are driven by the interplay between coagulation and inflammation. Inflammation drives atherosclerosis and IL-1α can play a dominant role independent of inflammasomes suggesting another mechanism activates IL-1α. Plaques contain thrombin-antithrombin complexes and show fibrin localized throughout, implying thrombin activation occurs throughout atherogenesis. Thus, p18 IL-1α might drive atherogenesis.’ 3
In super-short version:
Infection → inflammation + coagulation → (if regularly repeated) atherosclerotic plaques = cardiovascular disease
I find it a remarkable coincidence that I was studying the impact of infectious agents on cardiovascular disease when the COVID19 tsunami broke upon the world. Then I started delving into what the Sars-Cov2 virus does to a wide range of physiological systems. It opened doors into new passageways of thinking, and research, that I never even knew existed.
Primarily, that there is a tight connection between the blood clotting system and the immune system. Who knew? Well, some people obviously did, because they were researching it and writing about it. However, until COVID19 came along I didn’t have the faintest idea. I hadn’t even thought to connect the two processes.
Yes, I already knew that infectious diseases, such as Influenza, could greatly increase the risk of a fatal blood clot in the days and weeks following infection. I knew that sepsis (bacterial infection of the blood) causes damage to endothelial cells that line all blood vessels, triggering small blood clots all around the body. A condition known as Disseminated Intravascular Coagulation (DIC), which is the primary cause of death in sepsis.
I also knew that ‘inflammation’ of the blood vessels, a condition often known as vasculitis, could greatly increase the risk of cardiovascular disease. Vasculitis essentially means damage of the endothelium (the layer of glycocalyx, and endothelial cells, that line all blood vessel walls).
The impact of vasculitis on cardiovascular disease is highlighted by the fact that the form of vasculitis associated with Systemic Lupus Erythematosus (SLE) a.k.a. ‘lupus’ can increase the risk of death from cardiovascular disease by – up to – 4,900% in young women. 4
Indeed, all the vasculitides – plural of vasculitis – can greatly increase the risk of CVD, and thrombosis (blood clotting):
‘The relationship between inflammation and thrombosis is not a recent concept, but it has been largely investigated only in recent years. Nowadays inflammation-induced thrombosis is considered to be a feature of systemic autoimmune diseases such as Systemic Lupus Erythematosus (SLE), Rheumatoid Arthritis (RA), or Sjögren Syndrome (SS). Moreover, both venous and arterial thrombosis represents a well-known manifestation of Behçet syndrome (BS).’ 5
Then, of course, along comes COVID19, which brought a number of these strands into tight focus. It became clear that COVID19 also links infection + coagulation + vasculitis.
How so? Well, it was rapidly established that COVID19 enters cells by linking onto a receptor known as the ACE2 receptor (Angiotensin Converting Enzyme 2 receptor), before being dragged into the cell.
ACE2 receptors form an important part of the enormously complex Renin Aldosterone Angiotensin System (RAAS). Sorry, this is yet another strand, but please bear with me for a while, because it is important.
What is the Renin aldosterone angiotensin system? Well, keeping it super-simple, the RAAS controls blood pressure. If your blood pressure drops the RAAS kicks into action. [It also kicks into action if sodium levels fall, but that is an entirely different world of discussion]. The RAAS forces the heart to pump harder, it constricts blood vessels, it drives the kidneys to keep a hold of sodium and water etc. etc.
Although there are all sorts of hormones involved in the RAAS, with feedback and amplification loops here and there, they basically all end up triggering the conversion of a hormone called angiotensin I to angiotensin II. Angiotensin II is the active hormone that locks onto receptors in various organs, causing them to do their blood pressure raising thing.
[If you block the conversion of angiotensin I to angiotensin II, you will lower the blood pressure. This is what the class of drugs known as ACE-inhibitors do. They inhibit the enzyme that turns angiotensin I into angiotensin II. Which means that they are called angiotensin converting enzyme inhibitors. This reduces the amount of angiotensin II in the blood, and stops the heart rate increase, the blood vessel contraction, and suchlike. These drugs are widely prescribed]
As you might imagine therefore, ACE2 receptors are present in high numbers on the surface of membranes of cells that play a role in the RAAS. Basically, any cells involved in blood pressure control.
A large number are found in the cells in the lungs, because the lungs are where Angiotensin I (the inactive pro-hormone) is converted to Angiotensin II – the active form. Why does this conversion occur in the lungs, not the kidneys or liver? No idea. Something to do with evolution probably.
ACE2 receptors are also found in the cells that line all blood vessels – the endothelial cells. Why? Because angiotensin II links to these receptors to create messages commanding blood vessels to constrict – thus raising the blood pressure.
[In fact, sorry to add yet another complication, ACE2 receptors represent part of the ‘control feedback system’ for RAAS. When activated, ACE2 receptors block the effects of angiotensin II. They are ‘anti-angiotensin II’ receptors, if you like. They work to keep the effects of angiotensin II from running out of control. However, they are still an integral part of the RAAS system, and a critical part of the negative feedback loop to control blood pressure. Thus, wherever you have an ACE-receptor, you will also have an ACE2 receptor. Yin and Yang].
Why is all of this important, you may ask. Because it explains which cells are going to be most damaged by COVID19, and why. Essentially, the cells that are most damaged will be the cells that play a role in the RAAS. They are damaged because they have ACE2 receptors on their membranes.
Without this receptor, it is impossible for a cell to be infected by Sars-Cov2, and no damage can occur.
Years ago, I was looking at the Ebola virus. I found out that this virus gains entry through a protein stuck to the cell membrane known as the CCR5 protein. As with COVID19 and the ACE2 receptor, Ebola must find something on the cell membrane to link onto, before it can gain entry to the cell. A lock and key if you like. If the lock doesn’t fit the key – there can be no entry for the virus.
It was found that some people have a variant of this protein known as the ‘CCR5 Delta 32 mutation’. Because this protein has a different structure to the normal CCR5 protein, the Ebola virus cannot link to it. Therefore, it cannot enter any cells. Which means that people with the CCR5 Delta mutation cannot become infected with Ebola. Or at least, it cannot enter any cells in the body, so it cannot multiply, so it cannot cause any damage.
It is of interest that HIV also enters cells using the CCR5 protein, and people with the CCR5 delta 32 mutation cannot be infected with HIV either.
Anyway, trying desperately to bring things back together… deep breath. Once inhaled, COVID19 gets into lung cells using the ACE2 receptor – creating lung damage. It gets into kidney cells – creating further damage. It gets into heart cells (myocytes, pericytes) – causing even more damage. It gets into endothelial cells – creating vasculitis. It also stimulates the coagulation system into action – as almost all infectious agents do.
If you survive the initial lung damage – which most people probably will do – then the thing you need to start worrying about is the vasculitis/blood clotting that will be triggered throughout the rest of the body. This will all be worsened by the fact that infected endothelial cells will be sending out cytokines (distress messages) to the immune system. Stating, simply. ‘I am infected, come and kill me and the virions within.’
This, then, is the basis of the ‘cytokine storm’ which you may have read about with COVID19. Ironically, the body’s own defence system, the immune system, can become the very thing that kills you with COVID19. It revs up, starts attacking the infected cells, and creates major problems such as myocarditis (inflammation/damage to heart muscle). Kidney damage/failure, and a more widespread severe vasculitis develops as the endothelial cells are machine gunned by their own side.
All of this creates widespread blood clotting, which was recognised quite early on. Here from the paper ‘Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications.’
‘Reports of widespread thromboses and disseminated intravascular coagulation (DIC) in patients with coronavirus disease 19 (COVID-19) have been rapidly increasing in number. Key features of this disorder include a lack of bleeding risk, only mildly low platelet counts, elevated plasma fibrinogen levels, and detection of both severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and complement components in regions of thrombotic microangiopathy (TMA). This disorder is not typical DIC. Rather, it might be more similar to complement-mediated TMA syndromes, which are well known to rheumatologists who care for patients with severe systemic lupus erythematosus or catastrophic antiphospholipid syndrome.’ 6
Again, much jargon. However, the final sentence which provided me with the intellectual equivalent of sipping a twelve-year-old malt whisky… Roll it around the palate with deep pleasure. Please read again, and think about it:
‘Rather, it might be more similar to complement-mediated TMA syndromes, which are well known to rheumatologists who care for patients with severe systemic lupus erythematosus or catastrophic antiphospholipid syndrome.’
On the face of it, a rather boring sentence. What it is telling us, however, is that with COVID19 we are looking at almost the same pathological process as seen in Systemic Lupus Erythematosus (SLE), with an added dash of antiphospholipid syndrome.
Lupus, as mentioned before, causes vasculitis, because the immune system attacks endothelial cells. It is made worse when the person also has antiphospholipid syndrome (sometimes called Hughes’s syndrome).
Phospholipids essentially, are cell membranes. Two layers of phospholipids stuck back-to-back like Velcro. Within this bi-layer of phospholipids are various channels and gates and receptors and (as you may have noticed), lots of cholesterol – which stabilises the cell membrane. No cholesterol, no cell membrane, it simply falls apart.
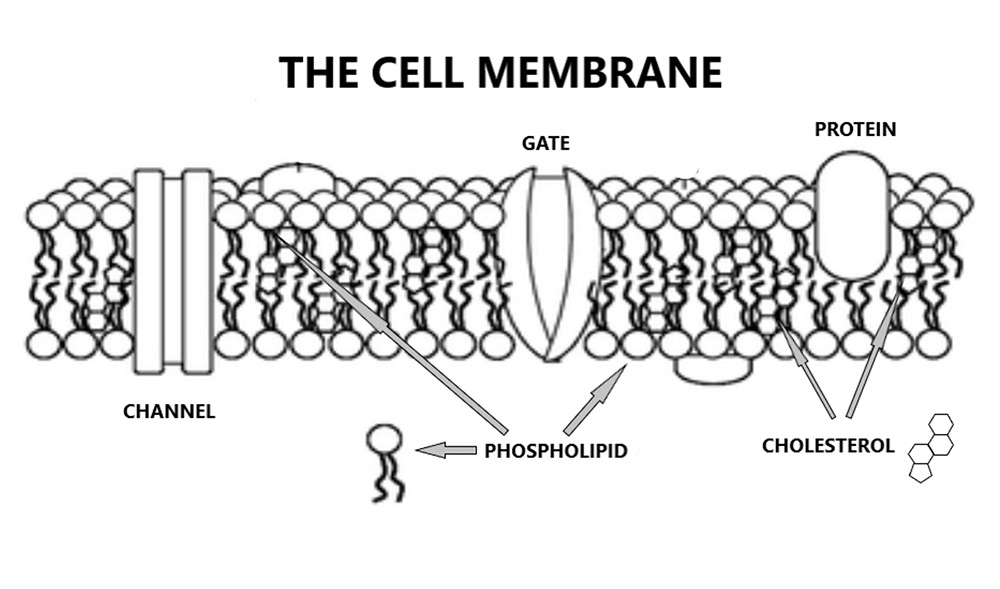
Getting back to anti-phospholipid syndrome, it means exactly what you would think it means. The immune system starts to attack the phospholipid bi-layer that makes up the endothelial cell membrane, it becomes an ‘anti-phospholipid system’. This creates damage, the damage exposes the underlying clotting factors, and you end up with blood clots forming on blood vessel walls. Thrombotic microangiopathy (TMA).
Thus SLE/antiphospholipid syndrome, and COVID19, although they are completely different diseases, can create almost the same damage. The immune system and clotting system combining – along with severe endothelial disruption. This is also, almost certainly, why some children develop a severe vasculitis following shortly after the acute phase of COVID19 infection.
Here, from the article ‘COVID-19-associated vasculitis and vasculopathy.’
‘COVID-19 is a SARS–CoV-2 syndrome that can involve all organs, including the circulatory system. Endothelial cell inflammation occurs within arteries, arterioles, capillaries, venules and veins and contributes to pathological events; including tissue hypoperfusion, injury, thrombosis and vascular dysfunction in the acute, subacute and possibly chronic stages of disease. Beyond re-writing the textbooks that hence will include SARS–CoV-2 as a causal pathogen for multi-bed vasculitis, the data will show that it is a new category of systemic vasculitis forever captured in the annals of medicine.’ https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7373848/
Look, I understand this is all complex, and I have taken you through it all at a bit of a rush, but I was hoping to give you a sense of my scientific excitement. When COVID19 hit, I was looking at vasculitis and how it caused cardiovascular disease. Here, are the very words I was writing.
‘Vasculitis means damage and inflammation to the blood vessels. Vascular = blood vessels; ‘itis’ = inflammation. As in tonsillitis = inflammation of the tonsils, or appendicitis = inflammation of the appendix.
There are many, many different sorts of vasculitis, and they all have impossible to remember names. However, I do love them, as they are so evocative of a bygone era in medicine. Here are several of them, not including systemic lupus erythematosus or rheumatoid arthritis:
- Polyarteritis nodosa
- Waldenström’s macroglobulinaemia
- Sjogren’s disease
- Giant cell arteritis
- Behcet’s disease
- Buerger’s disease
- Churg-Strauss syndrome
- Cryoglobulinemia
- Granulomatosis with polyangiitis
- Henoch-Schonlein purpura
- Kawasaki disease
- Takayasu’s arteritis
This is Harry Potter stuff. Wave your wand about and exclaim…’Vasculitis obliterans!’ Actually, that is another form of vasculitis. The reason why they don’t all appear on Qrisk3 is because many of them are considerably rarer than hen’s teeth. In addition, they not widely recognised to increase CVD risk – although they all do. If you choose to look.
Apart from increasing the risk of CVD, another characteristic they have in common is that they are also, what are termed as auto-immune conditions. ‘Autoimmune’ describes the situation whereby the body decides to attack itself….’
Immune system + vasculitis + coagulation.
How strange that a virus would come along and create an almost perfect model to highlight this world, I thought.
As a sign-off, I did wonder what it was with COVID19 that so directly stimulated the blood clotting system. As it turns out, it appears to be the spike protein itself. Here, from the paper ‘The unique characteristics of COVID-19 coagulopathy.’
‘Thrombosis is a major pathological driver in COVID-19. Evolving evidence suggests that in addition to the activated leukocytes and derangement of antithrombotic property of endothelial cells, hyperactive platelets participate in thrombogenesis. The direct and indirect effects of SARS-CoV-2 spike protein on platelets stimulate the release of platelet factor 4. The spike protein also upregulates inflammation and coagulation through the binding to ACE2 on macrophages/monocytes, lung epithelial cells, and possibly vascular endothelial cells, reactions that lead to micro and macro circulatory clotting known as CAC (COVID19 associated coagulopathy).’ 7
Yes, the spike protein. This, it appears, is the key antigen, the key driver of the immune/thrombotic system in COVID19. This is the factor that can lead to blood bloods, strokes heart attacks…sudden death.
‘The number of out-of-hospital sudden death episodes has increased since COVID-19 outbreaks. One of the possible reasons is the high incidence of major thrombotic events in patients with COVID-19.’
It would therefore seem that caution would be required, if you were to find a way to stimulate the creation of trillions of spike proteins within the human body. Caution.
Anyway, now you know – I hope – why I became so interested in COVID19. Because it links together a whole series of processes that, I believe, are key to understanding cardiovascular disease. Endothelial damage, blood clot formation, the central role of the blood clotting system.
Of course, COVID19 represents an acute vasculitis which comes and goes at some speed and is unlikely to lead to the longer-term damage required to create the repeated clot deposition necessary to drive atherosclerotic plaque formation. However, it can still cause acute clot formation, which can lead to strokes and heart attacks and kidney damage, and suchlike.
It is why, after I got vaccinated, I took aspirin for a month.
Next, fully back to cardiovascular disease – and associated stuff. I will even start to promote my new book – due to launch in October. ‘The enduring mystery of heart disease – The Clot Thickens.’ Yes, it was my son who came up with the title. Not that I will ever let anyone know it was him.
1:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6046589/
2: https://www.sciencedirect.com/science/article/pii/S1074761319300937
3: https://www.sciencedirect.com/science/article/pii/S1074761319300937
4: https://www.frontiersin.org/articles/10.3389/fmed.2018.00200/full
5: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4399148/
6: https://www.nature.com/articles/s41584-020-0474-5
7: https://ccforum.biomedcentral.com/articles/10.1186/s13054-020-03077-0












